

生物大分子三维空间结构数据库是一类重要的生物信息学数据库。蛋白质结构数据库(ProreinData Bank,PDB)是1971年创建的国际上最著名、最完整的蛋白质三维结构数据库。另外还有蛋白质分类数据库SCOP和CATH。SCOP是英国医学研究委员会分子生物学实验室和蛋白质工程中心开发的基于Web的蛋白质结构分类、检索和分析系统。CATH是另一个著名的蛋白质分类数据库,由英国伦敦大学开发和维护。1
理论依据20世纪60年代美国的Anfinsend小组作了牛胰核糖核酸酶变性及复现再折叠的实验和理论研究,提出蛋白质的氨基酸序列包含有足够的信息去决定它的空间结构(并因此获得诺贝尔奖),为由氨基酸序列预测蛋白质的三维空间结构建立了实验和理论基础。2
蛋白质结构蛋白质结构是发挥其生物学功能的基础,尽管实验方法可以解析出一部分蛋白质的高分辨率结构,但结构预测的理论方法依然非常重要,它将为大部分蛋白的结构研究提供非常有价值的信息,尤其是对那些不能从实验上测定结构的蛋白。实际上,精确的蛋白质三维结构理论预测是困难的,现阶段比较有效的途径是先研究粗粒化结构,如蛋白质结构型和拓扑结构等。由英国伦敦大学UCL开发和维护的蛋白质结构分类数据库CATH(Class—Architecture.Topology-Homology)、由英国医学研究委员会(MRC)的分子生物学实验室和蛋白质工程研究中心开发和维护蛋白质结构分类数据库SCOP(Structural Classification OfProteins)都是围绕粗粒化结构建立的。2
蛋白质结构预测应用PDB对PDB数据库库进行数据挖掘和统计的最终目的是为了能够通过对已知结构的蛋白质数据的数据挖掘,发现蛋白质序列和结构之间的某种联系或者规律,加深我们对蛋白质序列决定结构的机理的了解,并最终能寻找更好的蛋白质二级结构预测方法。3
发展历史世界上第一个蛋白质晶体结构的测定和解析发生在50年代末60年代初。蛋白质二级结构预测工作开始于60年代中期,也就是说在解析出第一个蛋白质的三维立体结构不久,科学家们便开始了蛋白质结构预测研究工作。这件事本身就可在一定程度上说明蛋白质结构预测工作的重要性。当时大多数的预测方法是依靠比对方法进行预测,而且直到目前为止对此问题还没有完全解决,往往是某种方法对一类或是几类蛋白质的二级结构的预测特别准确,而对其他几类蛋白质的预测却很不尽如人意。当然这其中有人们对整体蛋白质结构知识匮乏的因素,也有受限于当时的科学技术手段的因素。最早的比较成功的方法是Chou和Fastman在1974年提出了他们基于统计学的蛋白质二级交换结构预测的方法,之后又有很多人不断地在改进算法以期提高预测的准确度。这其中有1978年Gamier,1987年Deleage和Roux,1996年King和Sternberg的SOPMA算法,1999年Guermeur的DSC算法等等,近年来很多方法已经不仅仅基于统计学的方法,两是在原来统计学思想的基础上增加了多重序列比对、神经网络、SVM或是决策树的方法,其中最著名的就是1993年Rost和Sander的基于多重序列比对和多层神经网络的PHI)方法,蛋白质二级结构预测的准确度提高到70%,同时该方法也倡导了目前蛋自质二级结构预测中各种算法联用的趋势。3
基本方法生物信息学的主要目的之一在于了解蛋白质中氨基酸序列和三维结构之间的关系。如果知道了这种关系,就可以从氨基酸序列可靠地预测蛋白质结构。然而,序列和结构间的关系并不简单。
目前有超过160000条蛋白质的序列可用,但其中仅有35343条蛋白质的结构是已知的(PDB数据库2006-02.28的统计数据)。基因组的加速预计能加快解决蛋白质结构问题。蛋白质结构比较表明,新发现的蛋白质结构常常与已知结构具有相似的结构上的折叠或构造。因此,蛋白质折叠成三维结构的许多方法是一致的。结构比较亦显示出蛋白质中许多不同的序列在不同的结构环境中可以发现相同的短氨基酸模式。二级结构中的氨基酸序列也被收录在对结构预测有用的数据库中。序列数据库中许多蛋白质具有保守序列模式,这种保守模式可以进一步分类。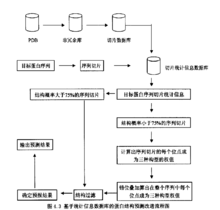
如果两个蛋白质序列显著相似,则它们也应有相似的三维结构。这种相似性可能体现在序列的长度上,或体现一个或多个已定位的区域中是否有相对短的模式。进行全局序列比对后,如果多于45%的氨基酸位置是相同的,则蛋白质三维结构中氨基酸的重叠性就高。因此,如果其中一个已比对过的蛋白质结构已知,则另一个蛋白质结构以及相同的氨基酸位置就可以可靠的预测。如果少于45%而多于25%的氨基酸相同,结构也可能相似,但在相应的三维位置上变异较多。为此,人们发展了大量的蛋白质结构预测方法。3
预测系统目前基于蛋白质统计信息数据的预测系统的Web Server正在筹建中,由于整个系统构建的工作量是相当大的,故而暂时没有发布。计划中的Web Server将提供以下服务:
1.跟踪PDB数据的数据更新,随时更新统计信息库中的数据;
2.提供蛋白质不同切片长度的结构概率及相关统计信息检索服务;
3.提供基于统计信息数据库的蛋白质二级结构预测服务。
随着生物信息的发展,人们必将获得越来越丰富的蛋白质结构数据,我们应该坚持利用数据挖掘这个有力的工具,对数据进行有效的处理,以期获得更多有益的知识。3
蛋白质综合信息数据库构建蛋白质信息数据库的建立是蛋白质结构研究的基础,围绕蛋白质的粗粒化结构及可能对蛋白质结构产生影响的因素,建立实用的非冗余的数据库是必要的。在非冗余结构分类数据库ASTRAL 的基础上,将蛋白质的序列信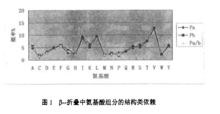 息、二级结构信息、蛋白质亚细胞定位信息、功能信息、物种信息、PDB ID信息、SWISS.PROT ID信息集成,得到蛋白质综合信息数据库。其中PDB ID、SWISS.PROT ID信息来自Swiss.Prot数据库,蛋白质序列信息、二级结构信息来PDB数据库,亚细胞定位信息来自DBSubLoc数据库,结构类信息来自SCOP数据库,蛋白质的物种信息信息取自PDBSOURCE,蛋白质的功能信息信息取自PDB RECORDS。2
息、二级结构信息、蛋白质亚细胞定位信息、功能信息、物种信息、PDB ID信息、SWISS.PROT ID信息集成,得到蛋白质综合信息数据库。其中PDB ID、SWISS.PROT ID信息来自Swiss.Prot数据库,蛋白质序列信息、二级结构信息来PDB数据库,亚细胞定位信息来自DBSubLoc数据库,结构类信息来自SCOP数据库,蛋白质的物种信息信息取自PDBSOURCE,蛋白质的功能信息信息取自PDB RECORDS。2
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国