

普伐他汀钠片,高脂血症、家族性高胆固醇血症。
成份本品主要成分为普伐他汀钠,其化学名称为:{1S-[1α(βS*,δS*),2α,6α,8β(R*),8aα]}-1,2,6,7,8,8a-六氢-β,δ,6-三羟-2-甲基-8-(2-甲基-1-氧丁氧基)-1-萘庚酸单钠盐 。
化学结构式:
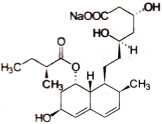
分子式:C23H35NaO7
性状本品为淡红色片。
适应症高脂血症、家族性高胆固醇血症。
规格(1)10mg、(2)20mg、(3)40mg。
用法用量成人开始剂量为10~20毫克,一日1次,临睡前服用。应随年龄及症状适宜增减,一日最高剂量40毫克。
不良反应总病例11,224例中,329例(2.93%,本项包括不能计算发生率的副作用)出现副作用(包括临床检验值异常),主要有皮疹(0.11%)、腹泻(0.08%)、胃部不适感(0.07%)等。
- 重大不良反应(发生率不详)
- 横纹肌溶解症:出现肌肉痛、乏力感、CPK上升、血中及尿中肌红蛋白上升为特征的横纹肌溶解症,随之引起急性肾功能衰竭等严重肾损害,若出现此类症状应立即停药。
- 肝功能障碍:可能出现伴有黄疸、显著AST及ALT上升等肝功能障碍,故应注意观察,此种情况应立即停药并给予适当处理。
- 血小板减少:可能出现血小板减少,故应注意观察,并采取适当的处理准备。(有伴有紫癜和皮下出血症状的血小板减少报告)。
- 肌病:有出现肌病的报告。
- 周围神经障碍:有出现周围神经障碍的报告。
- 过敏症状:有出现狼疮样综合征、血管炎等过敏症状的报告。
- 其他不良反应
禁忌对本品或本品中任何成份有过敏症既往史患者。
注意事项1. 与其他HMG-CoA还原酶抑制剂类似,本品可能升高碱性磷酸酶及转氨酶的水平。建议在治疗前,调整剂量前或其他需要时,应测定肝功能。伴有活动性肝脏疾病或不明原因的持续性转氨酶升高的患者,禁用本品。对近期患过肝脏疾病、提示有肝脏疾病(例如,不明原因的持续性转氨酶升高,黄疸)、酗酒的患者,需谨慎使用。对于这些患者,宜从最小推荐剂量开始,逐步调整到有效治疗剂量,并需密切观察。治疗期间,患者若出现转氨酶升高或者肝脏疾病的症状或体征,需肝功能复检,直到肝功能恢复正常。若AST或ALT持续超出正常值上限三倍或三倍以上,则停用本品。
2. 本品罕见引起横纹肌溶解伴继发于肌红蛋白尿的急性肾功能衰竭,可引起无并发症的肌痛。肌病表现为肌肉压痛或者关节附近肌无力,并有肌酸磷酸激酶(CPK)升高达正常上限的10倍以上。有弥散性肌痛、肌肉压痛或者肌无力,和/或CPK显著升高的患者,需考虑肌病的可能性。若出现肌肉疼痛、压痛或肌肉无力,特别是伴有乏力或发热,需立即向医生报告。如果出现CPK明显升高,怀疑有肌病或者确诊有肌病,停用本品。若患者出现急性或严重的会导致发生继发于横纹肌溶解的急性肾功能衰竭,如败血症、低血压、大手术、创伤;重症代谢性、内分泌疾病,电解质紊乱;未控制的癫痫等情况,暂停使用本品。当本品与氯贝特类药物合用时,临床上可能有肾功能异常,因此仅在临床确有必要时方可应用。
3. 下述患者应慎重用药:
⑴ 有严重肝损害或既往史患者。
⑵ 有严重肾损害或既往史患者。
⑶ 正在服用贝特类药物(苯扎贝特等)、免疫抑制剂(环孢素等)、烟酸的患者。
孕妇及哺乳期妇女用药1. 尚未确立妊娠期用药的安全性,因此孕妇或可能妊娠的妇女,仅在治疗的益处大于风险时方可给药。
2. 哺乳期妇女避免用药,不得已给药时,应停止哺乳。
儿童用药尚未确立小儿用药的安全性。
老年用药老年患者应考虑高龄引起肾功能降低的可能,应定期检查肾功能,观察患者症状,慎重给药。
药物相互作用经体内和体外实验证实,本品不经细胞色素P450 3A4代谢,因此不会与其他由细胞色素P450系统代谢的药物(如苯妥英钠、奎尼丁等)产生明显的相互作用,也不会与细胞色素P450 3A4抑制剂(如地尔硫卓、伊曲康唑、酮康唑、红霉素等)产生明显的相互作用。
华法令:华法林与普伐他汀钠40mg同时服用对凝血酶原时间不会产生影响。
西米替丁:普伐他汀钠单用或与西咪替丁合用时的普伐他汀0-l2小时的AUC之间没有区别。单用普伐他汀钠或普伐他汀钠与西米替汀合用的AUC与普伐他汀钠与抗酸药合用时的AUC具显著差异。
地高辛:0.2mg地高辛与20mg普伐他汀钠合用9天,地高辛的生物利用度未发生改变;普伐他汀的AUC有增高趋势,但普伐他汀与其代谢产物合并生物利用度没有发生改变。
环孢素:至今为止,已有一些环孢素与普伐他汀钠(剂量高至20mg)合用的临床资料,这些资料没有显示环孢素的浓度会受到普伐他汀的影响。
吉非贝齐:临床试验发现,普伐他汀钠与吉非贝齐合用,CPK水平升高和因骨骼肌肉症状而停药的发生率,与安慰剂对照组、单用吉非贝齐组、单用普伐他汀钠组相比,有升高的趋势,普伐他汀的尿排泄量及其蛋白结合均减少。建议普伐他汀钠不要和吉非贝齐联合使用。其他:与阿司匹林、抗酸剂(服用本品1小时后)、西咪替丁、烟酸合用药代动力学无明显差异。与利尿剂、抗高血压药、洋地黄、血管紧张素转换酶抑制剂、钙通道阻断剂、β-受体阻断剂及硝酸甘油合用无明显药物相互作用。
药物过量迄今在关于普伐他汀过量的报告中,未见明显临床症状与相关的临床实验室异常。如果发生过量服用,应该进行系统治疗,按要求建立支持性监测方法。
药理毒理1. 药理作用
本品为3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,选择性地作用于合成胆固醇的主要脏器肝脏和小肠,迅速且强力降低血清胆固醇值,改善血清脂质。
本品通过二方面发挥其降脂作用。第一为可逆性抑制HMG-CoA还原酶活性使细胞内胆固醇的量有一定程度的降低,导致细胞表面的低密度脂蛋白(LDL)受体数的增加,从而加强了由受体介导的低密度脂蛋白胆固醇(LDL-C)的分解代谢和血液中LDL-C的清除。第二,通过抑制LDL-C的前体----极低密度脂蛋白胆固醇(VLDL-C)在肝脏中的合成从而抑制LDL-C的生成。
研究表明总胆固醇(TC)、LDL-C及载脂蛋白B(Apo B)的升高可促使人体动脉粥样硬化的形成;同时,降低高密度脂蛋白胆固醇(HDL-C)与其转运复合物载脂蛋白A(Apo A)的水平,也与动脉粥样硬化形成相关。心血管患病率与死亡率随总胆固醇水平的升高而升高,随HDL水平的升高而降低。虽然甘油三酯水平的升高时常与低HDL水平伴随出现,但不能作为冠心病的独立风险因素。单纯升高HDL或降低甘油三酯对冠状动脉疾病与心血管疾病的发病或死亡率有何影响目前尚无定论,在健康志愿者与高胆固醇血症患者中,用本品治疗后可降低TC、LDL与Apo B,并降低VLDL和甘油三酯,升高HDL及Apo A,对其他诸如Lp(a)、纤维蛋白原等冠心病独立患病因素的影响效果尚不明确。临床研究表明,对伴有不同程度胆固醇升高的患者,本品能减少心血管疾病的发病率和死亡率。
2.毒理作用
SD系大鼠给于普伐他汀钠(混食给予10、30、100mg/千克/日,24个月)实验中,100mg/千克/日给药组(最大临床用量的250倍)中仅雄鼠发生的肝肿瘤较对照组明显,但雌鼠未发生。
狗给予普伐他汀钠(12.5、50、200mg/千克/日、5周、口服及12.5、50、200mg/千克/日、13周、口服)实验中,100mg/千克/日给药组见到脑微小血管渗出性出血等。
药代动力学1. 吸收、分布及代谢:本品为水溶性HMG-CoA还原酶抑制剂,主要从十二指肠吸收,口服后吸收迅速,高浓度分布于胆固醇生物合成旺盛的肝脏及小肠等,而在脑、肾上腺、生殖器等脏器的分布极低。本品给药后1-2小时即达最大血药浓度,血药浓度随给药量的增加而依存性增加。半衰期约为1.5小时,分布容积为830.0L,血清蛋白结合率为53.1%,AUC为14.0±3.9ng×hr/mL。本品主要经肝脏代谢,但不经细胞色素P450 3A4代谢,稳态AUCs、Cmax和Cmin分析均提示本品(普伐他汀)无论是每日一次或每日二次服用,体内都没有蓄积。
2. 排泄:本品通过肝、肾双通道进行清除:以粪中排泄为主(80%以上),尿中排泄为2%~13%。所以肾或肝功能不全的患者可通过代偿性改变排泄途径而清除。
贮藏遮光,密封保存。
包装铝塑包装,外加铝箔袋,7片/盒。
有效期36个月。
执行标准国家药品标准YBH00802004
1
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国