

氟伐他汀钠缓释片,适应症为用于饮食未能完全控制的原发性高胆固醇血症和混合型血脂异常(Fredrickson Ⅱa及Ⅱb型)的患者。
警示语本品会引起一定比例的患者肝脏转氨酶3倍以上的升高,在使用的过程中,应定期检查肝功能。如果出现谷丙转氨酶(ALT)或谷草转氨酶(AST)升高超过正常上限的3倍,应该停止治疗(详见【注意事项】)
成份活性成份:氟伐他汀钠。
化学名称:[R*,S*-(E)]-(±)- 7-[3-(4-氟苯基)-1-(1-甲基乙基)-1氢-吲哚-2-基]- 3,5-二羟-6-庚酸钠。
化学结构式:
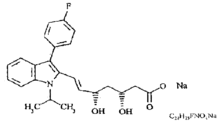
分子式:C24H25FNO4Na
分子量:433.46
性状本品为黄色圆形薄膜衣片,除去包衣后显淡黄色。
适应症用于饮食未能完全控制的原发性高胆固醇血症和混合型血脂异常(Fredrickson Ⅱa及Ⅱb型)的患者。
规格80毫克
用法用量成人患者
在开始治疗前,患者应采用标准的降低胆固醇的饮食控制。在整个治疗期间应继续饮食控制。
推荐的起始剂量为20mg或40mg常释胶囊,每日一次。可以根据患者治疗效果和推荐需达到的治疗目标调整剂量。对于严重的高胆固醇血症或者40mg常释胶囊治疗效果不满意的患者,可以使用氟伐他汀钠缓释片80mg/天。推荐的每日最大剂量为80mg。
本品可在每天任意时间服用,无论进食与否。给药后,四周内达到最大降脂作用。药物长期服用持续有效。
本品单独治疗有效。研究同时表明氟伐他汀和烟酸、消胆胺以及贝特类药物联合应用是安全和有效的(见【药物相互作用】)。
肾功能不全患者
氟伐他汀经肝脏清除,仅有不到6%的药物进入尿中。轻至中度肾功能不全的患者中氟伐他汀的药代动力学保持不变,无需调整剂量。
肝功能损害患者
活动性肝病或无法解释的血清转氨酶持续升高的患者,禁用本品(见【禁忌】及【注意事项】)。
不良反应药物不良反应在表1中按照medDRA系统器官分类列出。在每个系统器官分类中,根据其发生率对不良反应进行排列,最常见的不良反应排在首位。在每一个频率组中,将不良反应以严重性程度为序降序排列。此外,采用下列常规分类方法(CIOMS川分类方法)。对每项药物不良反应的发生率定义如下:非常常见(≥1/10);常见(≥1/100,3 x ULN,单次检测)异常。

全球试验数据
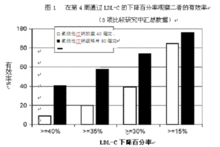
在3项多中心、双盲、活性药物对照的研究中,对近1700例原发性高胆固醇血症或原发性混合型血脂异常的患者,进行了氟伐他汀钠缓释片80毫克与氟伐他汀钠胶囊(睡前服40mg或40mg 每日二次)治疗24周的比较。达到最大疗效时,氟伐他汀钠胶囊(平均LDL-C下降26%)及氟伐他汀钠缓释片(平均LDL-C下降36%)的有效率见图1。
这些研究中在治疗24周之后,氟伐他汀钠胶囊及氟伐他汀钠缓释片二者都能显著降低TC、LDL-C、apo-B及TG,并升高HDL-C,呈剂量一致反应。(见表3)
表3 24周后与基线水平相比的平均变化百分率(所有患者)
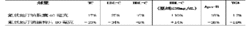
*平均变化的百分率
857例随机服用氟伐他汀钠缓释片的患者中,271例为原发性混合型血脂异常(FredricksonⅡb型)基线TG³200mg/dL,显示甘油三酯平均下降25%。同时,在这些患者中,氟伐他汀钠缓释片使HDL-C明显增加13%。在HDL-C基线水平很低的患者中(即[35mg/dL),这种效应尤为明显,这些患者平均HDL-C增加了16%,同时TC、LDL-C及Apo-B也出现了显著下降(见表4)。(这些研究中排除了TG]400mg/dL的患者。)

*平均变化的百分率
在脂蛋白和冠状动脉硬化研究中(Lipoprotein and Coronary Atherosclerosis Study, LCAS), 采用定量冠状动脉造影术评估了氟伐他汀对冠状动脉粥样硬化过程的影响。年龄35~75岁患轻-中度高胆固醇血症(基线LDL-C为115~190mg/dL)及冠状动脉粥样硬化性心脏病(CHD)的男女患者参加了这一随机双盲安慰剂对照的临床研究,429例患者给予氟伐他汀40mg/每天或安慰剂。在基线时和两年半时分别进行血管造影加以评估。结果显示氟伐他汀治疗延缓了患者冠状动脉粥样硬化病变的进展,2.5年后最小管腔直径增加了0.07 mm(95%可信区间,从-0.1222到-0.022)的变化。因此可将氟伐他汀钠用于延缓原发性高胆固醇血症患者的冠状动脉粥样硬化的进展,包括轻度动脉粥样硬化和冠心病。
氟伐他汀钠干预预防研究(Lescol Intervention Prevention Study, LIPS)证实了氟伐他汀在冠状动脉导管治疗后对主要不良心血管事件的二级预防作用。参加研究的冠心病患者年龄18-80岁,基础胆固醇水平3.5-7.0 mmol/L。 这是一项随机、双盲、安慰剂对照试验,给予氟伐他汀(N=844)每天80 mg 治疗4年,与对照组(N=833)相比,氟伐他汀组显著减少了首次主要心血管事件(MACE)达22%(p=0.013)。特别是在糖尿病和多支血管病变的患者中,这种益处更为突出。氟伐他汀可以显著降低心源性死亡和/或心肌梗死的危险达31%(p=0.065),因此可将氟伐他汀钠用于冠状动脉导管治疗后的二级预防,减少主要不良心血管事件的发生(心源性死亡,非致死性心肌梗死和冠状动脉重建)。
儿童患者
两项为期2年的开放性,剂量递增的试验(ZA01和2301)研究氟伐他汀20mg至80mg的安全性和有效性,总共有113例杂合子家族性高胆固醇血症的儿童和青少年患者参加。
试验入组的9岁及9岁以上的杂合子家族性高胆固醇血症患者应符合以下入选标准:
· LDL-C 水平≥ 190 mg/dL (4.9 mmol/L)
· 或 LDL-C水平 ≥ 160 mg/dL (4.1 mmol/L) 并伴有一个或多个危险因素(早发冠心病的家族病史、吸烟、高血压、高密度脂蛋白胆固醇 (HDL-C) · 或已证实有LDL-C受体脱氧核糖核酸 (DNA) 缺陷,且LDL-C水平>160 mg/dL (4.1 mmol/L),血浆中甘油三酯水平为 600 mg/dL或更低。
主要的排除标准为:同合子家族性高胆固醇血症患者;异常脂蛋白血症(II型);血浆甘油三酯水平>600 mg/dL;ALAT, ASAT 或肌酐水平> 1.5 x ULN;血浆 CK或血浆TSH> 2 x ULN;BMI > 30 kg/m2。
第一周氟伐他汀的起始治疗剂量为20 mg,逐渐增加为40 mg(以6周为间隔)。如果 LDL-C 水平] 3.2 mmol/L 或 3.4 mmol/L,则增加到80mg (2倍40mg氟伐他汀钠胶囊或者80mg氟伐他汀钠缓释片)。
通过2年的随访发现,氟伐他汀可以显著降低血浆中TC, LDL-C, TG 以及Apo- B水平 ,并可升高HDL-C 水平(结果见表 5)。
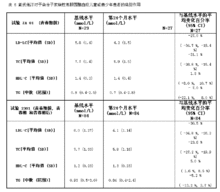
在这两项试验中,所有患者的生长和性成熟均未受影响。对年龄小于9岁的患者尚未进行氟伐他汀的研究。
这些试验结果不能推断在儿童患者早期进行降脂治疗对心血管事件的益处。
药理毒理药物分类:HMG-CoA还原酶抑制剂。
氟伐他汀是一个全合成的降胆固醇药物,是羟甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,可与HMG-CoA还原酶竞争,抑制HMG-CoA转化为3-甲基-3,5-二羟戊酸,而3-甲基-3,5-二羟戊酸是甾醇包括胆固醇的前体物质。本品的作用部位主要在肝脏,具有抑制内源性胆固醇的合成,降低肝细胞内胆固醇的含量,刺激低密度脂蛋白(LDL)受体的合成,提高LDL微粒的摄取,降低血浆总胆固醇浓度的作用。
在高胆固醇血症和混合性血脂紊乱的患者中,本品可以减少总胆固醇(TC),低密度脂蛋白胆固醇(LDL-C),载脂蛋白B(apo-B)和甘油三酯(TG)水平,增加高密度脂蛋白胆固醇(HDL-C)水平。在使用药物2周内出现良好的治疗效果,从治疗开始4周之内出现最大的效应,此效应在整个治疗过程中持续存在。
临床前安全性资料
急性毒性
氟伐他汀在小鼠的LD50大于2 g/kg,在大鼠的LD50大于0.7 g/kg。
重复剂量毒性
在大鼠、兔子、狗、猴子、小鼠以及仓鼠中进行了深入的毒性研究,评价氟伐他汀的安全性。研究结果与HMG-CoA还原酶抑制剂的结果相同,例如啮齿类动物非腺型胃部的细胞增生和过度角化,狗出现白内障,啮齿类动物出现肌病,大多数试验动物的肝脏出现轻微的变化,狗、仓鼠和猴子的胆囊出现变化,大鼠甲状腺重量增加,仓鼠睾丸出现病变。尚无试验数据表明,氟伐他汀和其它此类药物对狗CNS血管及变性方面有所影响。
致癌性
以每天6、9、18 mg/kg的给药剂量(一年后将剂量增加到每天24 mg/kg)进行的一项大鼠致癌性研究,以获得最大耐受剂量。在这样的给药剂量下,相当于人治疗剂量(口服给药40 mg)下血药浓度的9、13和26-35倍。在每天24 mg/kg的剂量水平下,出现胃前鳞状细胞乳头状瘤和胃前癌的机率非常低。此外,对于每天按18-24 mg/kg给药的雄性大鼠,甲状腺滤泡细胞瘤以及甲状腺滤泡细胞癌的发病率有所升高。
以每天0.3、15、30 mg/kg的给药剂量进行小鼠致癌性研究,与大鼠致癌性研究一样,在每天30 mg/kg剂量下的雄性和雌性小鼠以及在每天15 mg/kg的雌性小鼠,其胃前鳞状细胞乳头状瘤的机率明显增加。在这样的给药剂量下,相当于人治疗剂量(口服给药40 mg)下血药浓度的0.2、10和21倍。
大鼠和小鼠出现胃前肿瘤是由于动物的胃部长期与氟伐他汀直接接触而导致细胞增生,而不是药物具有致癌作用。给予氟伐他汀后,大鼠甲状腺滤泡细胞瘤发生率有所增加,这与其他HMG-CoA还原酶抑制剂的试验结果相同。与其他HMG-CoA还原酶抑制剂不同的是,氟伐他汀钠并未导致肝脏肿瘤发生率增加。
致突变性
通过体外试验发现,无论是否存在大鼠肝脏代谢活性,氟伐他汀均没有出现致突变性。主要试验包括:采用鼠伤寒沙门氏菌或大肠杆菌的突变菌株进行的微生物突变试验;BALB/3T3的恶性变异分析;大鼠肝细胞的异常DNA合成;V79中国仓鼠细胞染色体异常;HGPRT V79中国仓鼠细胞等。并且,氟伐他汀在大鼠或小鼠体外试验中没有出现致突变性。
生殖毒性
在一项生殖毒性动物试验中,雌性大鼠的给药剂量分别为每天0.6、2、6 mg/kg,雄性大鼠的给药剂量分别是2、10、20 mg/kg,氟伐他汀没有出现任何生殖毒性。在一项对大鼠(1、12以及36 mg/kg)和兔子(0.05、1以及10 mg/kg)进行的致畸试验中,结果显示氟伐他汀在高剂量下具有母代毒性,但并不具有胚胎毒性和致畸作用。在一项对雌性大鼠进行的试验中,在大鼠怀孕后期到断奶分别按每天12和24 mg/kg的剂量给予氟伐他汀,结果导致在产前和产后的母体死亡,并伴有子代死亡。在每天2 mg/kg低剂量给药下,对母代和子代均没有影响。
另一项试验是在怀孕晚期和哺乳期早期的给药剂量分别为每天2、6、12以及24 mg/kg,试验结果与前一个试验相似。在第三项试验中,在怀孕晚期至断乳期间,分别在同时给予3-甲基-3,5-二羟戊酸或不给予3-甲基-3,5-二羟戊酸的条件下按每天12和24 mg/kg剂量给予氟伐他汀,3-甲基-3,5-二羟戊酸为HMG-CoA的衍生物,对于胆固醇的生物合成有重要的影响。同时给予3-甲基-3,5-二羟戊酸完全可预防心脏毒性和子代及母代的死亡率。因此,母代和子代的死亡是氟伐他汀在孕期内药理作用增强的结果。
药代动力学吸收
健康志愿者空腹服用氟伐他汀溶液后,吸收迅速(98%)。在口服氟伐他汀钠缓释片后,与胶囊相比,氟伐他汀的吸收率延缓了60%,而平均体内留存时间延长了将近4小时。如果在进食的条件下,药物的吸收率有所降低。
分布
氟伐他汀主要作用于肝脏,肝脏也是其代谢的主要器官。从全身血药浓度计算其绝对生物利用度为24%。氟伐他汀的表观分布容积(Vz/f)为330升。超过98%的循环药物与血浆蛋白结合,这种结合与氟伐他汀,华法令,水杨酸和格列苯脲的血药浓度无关。
代谢
氟伐他汀主要在肝脏中代谢。循环药物主要为氟伐他汀原型和无药理学活性的代谢产物N-去异丙基丙酸。羟化的代谢产物有药理学活性,但不进入全身血液循环。氟伐他汀在人类肝脏的代谢途径已经被阐明,氟伐他汀的生物转化是通过多种细胞色素P450(CYP450)途径完成的,抑制细胞色素P450对氟伐他汀的代谢影响很小,而对细胞色素P450的抑制是药物相互作用的主要原因。
一些体外试验研究了氟伐他汀对细胞色素P450同工酶的抑制作用。氟伐他汀只会抑制通过CYP2C9代谢的化合物的代谢。这提示了氟伐他汀和以CYP2C9为作用底物的化合物可能存在竞争性的相互作用,这些药物包括双氯酚、苯妥英、甲苯磺丁脲和华法令,但临床上尚无数据证实。
消除
健康志愿者服用3H标记的氟伐他汀后大约6%的放射性活性出现在尿中,93%在粪便中,排出的氟伐他汀只占不到标记总量的2%。氟伐他汀的血浆清除率(CL/f)为1.8±0.8 L/min。服用氟伐他汀80mg/日的稳态血浆浓度未显示蓄积效应。
氟伐他汀和晚餐同时服用与在晚餐后4小时服用的AUC无明显不同。
特殊情况下的药代动力学
在一般人群中,氟伐他汀的血浆浓度并不随年龄和性别而改变。但是,在妇女和老人中发现对其治疗反应增强。
因为氟伐他汀主要经胆汁排泄并且进入体循环前有显著的生物转化,因此在肝功能不全的患者不能排除出现药物蓄积的可能性。(见【药物相互作用】和【注意事项】)
贮藏密封,25℃以下保存。
包装双铝包装,每盒7、14,28片
有效期24个月
执行标准国家食品药品监督管理局标准YBH041520091
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国