

克拉霉素片,本品适用于克拉霉素敏感菌所引起的下列感染:1.鼻咽感染:扁桃体炎、咽炎、副鼻窦炎;2.下呼吸道感染:包括支气管炎、细菌性肺炎、非典型肺炎;3.皮肤感染,脓疱病、丹毒、毛囊炎、疖和伤口感染。
成份克拉霉素。辅料为:低取代羟丙基纤维素、羧甲基淀粉钠等。
化学名称:6-O-甲基红霉素。
化学结构式:
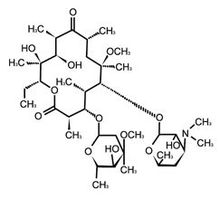
分子式:C38H69NO13
分子量:747.96
性状本品为薄膜衣片,除去包衣后显白色或类白色。
适应症本品适用于克拉霉素敏感菌所引起的下列感染:
1.鼻咽感染:扁桃体炎、咽炎、副鼻窦炎;
2.下呼吸道感染:包括支气管炎、细菌性肺炎、非典型肺炎;
3.皮肤感染,脓疱病、丹毒、毛囊炎、疖和伤口感染。
规格0.25g
用法用量口服,成人常用量每次0.25g,每12小时一次;重症感染者每次0.5g,每12小时一次。或遵医嘱。
不良反应1.主要有口腔异味(3%),腹痛、腹泻、恶心、呕吐等胃肠道反应(2%~3%),头痛(2%),血清氨基转移酶短暂升高。
2.可能发生过敏反应,轻者为药疹、荨麻疹,重者为过敏及Stevens—Johnson症。
3.偶见肝毒性、艰难梭菌引起的假膜性肠炎。
4.曾有发生短暂性中枢神经系统不良反应的报告,包括焦虑、头昏、失眠、幻觉、恶梦或意识模糊,然而其原因和药物的关系仍不清楚。
禁忌1.对本品或大环内酯类药物过敏者禁用。
2.孕妇、哺乳期妇女禁用。
3.严重肝功能损害者、水电解质紊乱患者、服用特非那丁治疗者禁用。
4.某些心脏病(包括心律失常、心动过缓、Q-T间期延长、缺血性心脏病、充血性心力衰竭等)患者禁用。
注意事项1.肝功能损害、中度至严重肾功能损害者慎用。
2.肾功能严重损害(肌酐清除率小于30ml/分钟)者,须作剂量调整。常用量为一次0.25g,一日1次;重症感染者首剂0.5g,以后一次0.25g,一日2次。
3.本品与红霉素及其他大环内酯类药物之间有交叉过敏和交叉耐药性。
4.与别的抗生素一样,可能会出现真菌或耐药细菌导致的严重感染,此时需要中止使用本品,同时采用适当的治疗。
5.本品可空腹口服,也可与食物或牛奶同服,与食物同服不影响其吸收。
6.血液或腹膜透析不能降低本品的血药浓度。
7.实验动物中克拉霉素对胚胎及胎儿有毒性作用。本品属妊娠期用药C类,克拉霉素及其代谢产物可进入母乳中,孕妇、乳妇患者需用本品时应充分权衡利弊后决定是否应用。如确有应用指征的乳妇患者应用本品时宜暂停授乳。
8.药物不要放在孩童可触及的地方。
9.废弃药品包装不应随意丢弃。
孕妇及哺乳期妇女用药动物实验中本品对胚胎及胎儿有毒性作用,同时本品及其代谢物可进入母乳中,故孕妇及哺乳期妇女禁用。
儿童用药6个月以下儿童的疗效和安全性尚未确定。
老年用药老年人的耐受性与年轻人相仿。
药物相互作用1.本品可轻度升高卡马西平的血药浓度,两者合用时需对后者作血药浓度监测。
2.本品对氨茶碱、茶碱的体内代谢略有影响,一般不需要调整后者的剂量,但氨茶碱、茶碱应用剂量偏大时需监测血浓度。
3.与其他大环内酯类抗生素相似,本品会升高需要经过细胞色素P450系统代谢的药物的血清浓度(如阿司咪唑、华法林、麦角生物碱、三唑仑、咪达唑仑、环孢素、奥美拉唑、雷尼替丁、苯妥因、溴隐亭、阿芬他尼、海索比妥、丙吡胺、洛伐他汀、他克莫司等)。
4.与HMG—CoA还原酶抑制药(如洛伐他汀和辛伐他汀)合用,极少有横纹肌溶解的报道。
5.与西沙必利、匹莫齐特合用会升高后者血浓度,导致Q—T间期延长,心率失常如室性心动过速、室颤和充血性心力衰竭。与阿司咪唑合用会导致Q—T间期延长,但无任何临床症状。
6.大环内酯类抗生素能改变特非那丁的代谢而升高其血浓度,导致心律失常如室性心动过速、室颤和充血性心力衰竭。
7.与地高辛合用会引起地高辛血浓度升高,应进行血药浓度监测。
8.HIV感染的成年人同时口服本品和齐多夫定时,本品会干扰后者的吸收,使其稳态血浓度下降,应错开服用时间。
9.与利托那韦合用,本品代谢会明显被抑制,故本品每天剂量大于1g 时,不应与利托那韦合用。
10.与氟康唑合用会增加本品血浓度。
药物过量当服用大剂量的克拉霉素时,可能有胃肠不适。因过量引起症状时应迅速洗胃并适当给予支持疗法。
药理毒理1.药理 本品为大环内酯类抗生素,对革兰阳性菌如金黄色葡萄球菌、链球菌、肺炎球菌等有抑制作用,对部分革兰阴性菌如流感嗜血杆菌、百日咳杆菌、淋病奈瑟菌、嗜肺军团菌和部分厌氧菌如脆弱拟杆菌、消化链球菌、痤疮丙酸杆菌等也有抑制作用,此外对支原体也有抑制作用。本品特点为在体外的抗菌活性与红霉素相似,但在体内对部分细菌如金黄色葡萄球菌、链球菌、流感嗜血杆菌等的抗菌活性比红霉素强。本品与红霉素之间有交叉耐药性。本品的作用机制是通过阻碍细胞核蛋白50S亚基的联结,抑制蛋白合成而产生抑菌作用。
2.毒理 本品除体外染色体畸变试验一次是弱阳性、另一次为阴性结果外,其他体外致突变试验如沙门菌/哺乳动物细胞微粒体试验、细菌致突变频率试验、大鼠肝细胞DNA合成测定、小鼠淋巴瘤测定、小鼠显性致死试验和小鼠微核试验均为阴性。生育、生殖研究表明,每日给雌性、雄性大鼠用克拉霉素160mg/kg(以体表面积计,是人用最大推荐剂量的1.3倍),对大鼠动情期、生育能力、分娩及子代的数量和存活率均无影响。每日给予猴子口服克拉霉素150mg/kg (以体表面积计,是人用最大推荐剂量的2.4倍),出现胚胎丧失。动物长期毒性研究未证实克拉霉素有致癌性。
药代动力学口服后经胃肠道迅速吸收,生物利用度(F)为55%。食物可稍延缓吸收,但不影响生物利用度。单剂口服400mg后2.7小时达血药峰浓度(Cmax)2.2mg/L;每12小时口服250mg,在2~3天内达到稳态血浓度约为1mg/L,其代谢物(14—羟基克拉霉素)为0.6mg/L,每12小时口服500mg,药物在稳定峰值状态的血浆浓度平均为2.7~2.9 mg/L,其代谢物为0.83~0.88 mg/L。体内分布广泛,鼻粘膜、扁桃体及肺组织中的药物浓度比血浓度高。在血浆中,蛋白结合率为65%~75%。其主要代谢物是具有大环内酯类活性作用的14—羟基克拉霉素。单剂给药后血消除半衰期(t1/2β)为4.4小时;每12小时口服250mg后的原形药物血消除半衰期(t1/2β)为3~4小时,其代谢物为5~6小时;每12小时口服500mg后的原形药物的血消除半衰期(t1/2β)为4.5~4.8小时,其代谢物为 6.9~8.7小时。经口服或静脉注入14C标记的克拉霉素,5日内自尿排出占剂量的36%,自大便排出占52%。低剂量给药经粪、尿两个途径排出的药量相仿,但剂量增大时尿中排出量较多。
贮藏遮光,密封,在阴凉干燥处(不超过20℃)保存。
包装包装材料:铝箔+聚氯乙烯固体药用硬片;
包装规格:每板6片,每盒1板。
有效期24个月
执行标准《中国药典》2010年版二部
1
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国