

分子动力学是一门结合物理,数学和化学的综合技术。分子动力学是一套分子模拟方法,该方法主要是依靠牛顿力学来模拟分子体系的运动,以在由分子体系的不同状态构成的系统中抽取样本,从而计算体系的构型积分,并以构型积分的结果为基础进一步计算体系的热力学量和其他宏观性质。
简史1980年:恒压条件下的动力学方法
1983年:非平衡态动力学方法(Gillan and Dixon)
1984年:恒温条件下的动力学方法
1985年:第一原理分子动力学法
1991年:巨正则系综的分子动力学方法(Cagin and Pettit)
基本步骤起始构型进行分子动力学模拟的第一步是确定起始构型,一个能量较低的起始构型是进行分子模拟的基础,一般分子的起始构型主要来自实验数据或量子化学计算。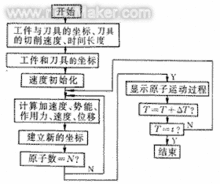 在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据波尔兹曼分布随机生成的,由于速度的分布符合波尔兹曼统计,因此在这个阶段,体系的温度是恒定的。另外,在随机生成各个原子的运动速度之后须 进 行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。
在确定起始构型之后要赋予构成分子的各个原子速度,这一速度是根据波尔兹曼分布随机生成的,由于速度的分布符合波尔兹曼统计,因此在这个阶段,体系的温度是恒定的。另外,在随机生成各个原子的运动速度之后须 进 行调整,使得体系总体在各个方向上的动量之和为零,即保证体系没有平动位移。
平衡相由上 一步 确定的分子组建平衡相,在构建平衡相的时候会对构型、温度等参数加以监控。
生产相进入生产相之后体系中的分子和分子中的原子开始根据初始速度运动,可以想象其间会发生吸引、排斥乃至碰撞,这时就根据牛顿力学和预先给定的粒子间相互作用势来对各个粒子的运动轨迹进行 计算 ,在这个过程中,体系总能量不变,但分子内部势能和动能不断相互转化,从而 体系的 温度也不断变化,在整个过程中,体系会遍历势能面上的各个点,计算的样本正是在这个过程中抽取的。 +
计算结果用抽样所得体系的各个状态计算当时体系的势能,进而计算构型积分。 作用势与动力学计算
作用势与动力学计算
作用势的选择与动力学计算的关系极为密切,选择不同的作用势,体系的势能面会有不同的形状,动力学计算所得的分子运动 和 分子内部运动的轨迹也会不同,进而影响到抽样的结果和抽样结果的势能计算,在计算宏观体积和微观成分关系的时候主要采用刚球模型的二体势,计算系统能量,熵等关系时早期多采用Lennard-Jones、morse势等双体势模型,对于金属计算,主要采用morse势,但是由于通过实验拟合的对势容易导致柯西关系,与实验不符,因此在后来的模拟中有人提出采用EAM等多体势模型,或者采用第一性原理计算结果通过一定的物理方法来拟合二体势函数。但是相对于二体势模型,多体势往往缺乏明确的表达式,参量很多,模拟收敛速度很慢,给应用带来很大的困难,因此在一般应用中,通过第一性原理计算结果拟合势函数的L-J,morse等势模型的应用仍然非常广泛。
时间步长与约束动力学分子动力学计算的基本思想是赋予分子体系初始运动状态之后利用分子的自然运动在相空间中抽取样本进行统计计算,时间步长就是抽样的间隔,因而时间步长的选取对动力学模拟非常重要。太长的时间步长会造成分子间的激烈碰撞,体系数据溢出;太短的时间步长会降低模拟过程搜索相空间的能力,因此一般选取的时间步长为体系各个自由度中最短运动周期的十分之一。 但是通常情况下,体系各自由度中运动周期最短的是各个化学键的振动,而这种运动对计算某些 宏观性质 并不产生影响,因此就产生了屏蔽分子内部振动或其他无关运动的约束动力学,约束动力学可以有效地增长分子动力学模拟的时间步长,提高搜索相空间的能力。
但是通常情况下,体系各自由度中运动周期最短的是各个化学键的振动,而这种运动对计算某些 宏观性质 并不产生影响,因此就产生了屏蔽分子内部振动或其他无关运动的约束动力学,约束动力学可以有效地增长分子动力学模拟的时间步长,提高搜索相空间的能力。
步骤以下是做模拟的一般性步骤,具体的步骤和过程依赖于确定的系统或者是软件,但这不影响我们把它当成一个入门指南:
1)首先我们需要对我们所要模拟的系统做一个简单的评估, 三个问题是我们必须要明确的:
做什么(what to do)为什么做(why to do)怎么做(how to do)
2)选择合适的模拟工具,大前提是它能够实现你所感兴趣的目标,这需要你非常谨慎的查阅文献,看看别人用这个工具都做了些什么,有没有和你相关的,千万不要做到一半才发现原来这个工具根本就不能实现你所感兴趣的idea,切记!
考虑1:软件的选择,这通常和软件主流使用的力场有关,而软件本身就具体一定的偏向性,比如说,做蛋白体系,Gromacs,Amber,Namd均可;做DNA, RNA体系,首选肯定是Amber;做界面体系,Dl_POLY比较强大,另外做材料体系,Lammps会是一个不错的选择
考虑2:力场的选择。力场是来描述体系中最小单元间的相互作用的,是用量化等方法计算拟合后生成的经验式,有人会嫌它粗糙,但是它确确实实给我们模拟大系统提供了可能,只能说关注的切入点不同罢了。常见的有三类力场:全原子力场,联合力场,粗粒化力场;当然还有所谓第一代,第二代,第三代力场的说法,这里就不一一列举了。
再次提醒注意:必须选择适合于我们所关注体系和我们所感兴趣的性质及现象的力场。
3)通过实验数据或者是某些工具得到体系内的每一个分子的初始结构坐标文件,之后,我们需要按我们的想法把这些分子按照一定的规则或是随机的排列在一起,从而得到整个系统的初始结构,这也是我们模拟的输入文件。
4)结构输入文件得到了,我们还需要力场参数输入文件,也就是针对我们系统的力场文件,这通常由所选用的力场决定,比如键参数和非键参数等势能函数的输入参数。
5)体系的大小通常由你所选用的box大小决定,我们必须对可行性与合理性做出评估,从而确定体系的大小,这依赖于具体的体系,这里不细说了。 6)由于初始构象可能会存在两个原子挨的太近的情况(称之为bad contact),所以需要在正式模拟开始的第一步进行体系能量最小化,比较常用的能量最小化有两种,最速下降法和共轭梯度法,最速下降法是快速移除体系内应力的好方法,但是接近能量极小点时收敛比较慢,而共轭梯度法在能量极小点附近收敛相对效率高一些,所有我们一般做能量最小化都是在最速下降法优化完之后再用共轭梯度法优化,这样做能有效的保证后续模拟的进行。
6)由于初始构象可能会存在两个原子挨的太近的情况(称之为bad contact),所以需要在正式模拟开始的第一步进行体系能量最小化,比较常用的能量最小化有两种,最速下降法和共轭梯度法,最速下降法是快速移除体系内应力的好方法,但是接近能量极小点时收敛比较慢,而共轭梯度法在能量极小点附近收敛相对效率高一些,所有我们一般做能量最小化都是在最速下降法优化完之后再用共轭梯度法优化,这样做能有效的保证后续模拟的进行。
7)以平衡态模拟为例,你需要设置适当的模拟参数,并且保证这些参数设置和力场的产生相一致,举个简单的例子,gromos力场是用的范德华势双截断来定范德华参数的,若你也用gromos力场的话也应该用双截断来处理范德华相互作用。常见的模拟思路是,先在NVT下约束住你的溶质(剂)做限制性模拟,这是一个升温的过程,当温度达到你的设定后, 接着做NPT模拟,此过程将调整体系的压强进而使体系密度收敛。
经过一段时间的平衡模拟,在确定系统弛豫已经完全消除之后,就可以开始取数据了。如何判断体系达到平衡,这个问题是比较技术性的问题,简单的讲可以通过以下几种方式,一,看能量(势能,动能和总能)是否收敛;二,看系统的压强,密度等等是否收敛;三看系统的RMSD是否达到你能接受的范围,等等。
8)运行足够长时间的模拟以确定我们所感兴趣的现象或是性质能够被观测到,并且务必确保此现象出现的可重复性。
9)数据拿到手后,很容易通过一些可视化软件得到轨迹动画,但这并不能拿来发文章。真正的工作才刚刚开始——分析数据,你所感兴趣的现象或性质只是表面,隐含在它们之中的机理才是文章中的主题。
应用分子动力学可以用于NPT,NVE,NVT等系综的计算,是一种基于牛顿力学确定论的热力学计算方法,与蒙特卡洛法相比在宏观性质计算上具有更高的准确度和有效性,可以广泛应用于物理,化学,生物,材料,医学等各个领域。
发展方向分子动力学模拟是研究微观世界的有效手段"其势函数和数值算法对模拟的精度有较大影响,为了提高势函数的精确性,将基于局部密度泛涵理论的从头计算分子动力学, 量子化学分析参数拟合和蒙特卡洛方法相结合有望成为研究势函数的最佳方法,随着计算机性能的不断提高,摆脱了经验势函数的从头计算分子动力学的应用范围将会不断扩大,计算的精度也会不断提高。所以,从头计算分子动力学将会成为分子动力学模拟未来的主要发展方向。
数值算法的高速和高效也是人们一直奋斗的目标,最近有人提出的多重时间宽度法,由于有效地减少了计算时间而可能成为分子动力学方法中较有前途的数值积分算法,分子动力学方法与其他计算方法 ,如有限单元法、 模拟淬火法、 蒙特卡罗法等的结合也将成为未来的发展方向之一1。
本词条内容贡献者为:
王强 - 副教授 - 西南大学
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国