

范围
本标准规定了原粮和成品粮中各项卫生指标的分析方法。
镉、氟、曼陀罗籽、麦角、二溴乙烷、七氯、艾氏剂、狄氏剂等卫生指标的分析。2
规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用予本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。
GB/T 5009.11 食品中总砷及无机砷的测定
GB/T 5009.15 食品中锅的测定
GB/T 5009.17 食品中总汞及甲基汞的测定
GB/T 5009.18 食品中氟的测定
GB/T 5009.19 食品中六六六、滴滴涕残留量的测定
GB/T 5009.20 食品中有机磷农药残留量的测定
GB/T 5009.22 食品中黄曲霉毒素的测定
感官检查具有正常粮食的色泽及气味,不得有发霉变质现象。
理化检验马拉硫磷气相色谱法
按GB/T 5009.20操作。
铜络合物比色法
原理
马拉硫磷用有机溶剂提取,经氢氧化钠水解后,生成二甲基二硫代磷酸酯,再与铜盐生成黄色络合物,与标准系列比较定量。
本方法取样量为20 g时,检出限为1.25 mg/kg。
试剂
四氯化碳。
无水乙醇。
硫酸钠溶液(45 g/L):称取4.5 9无水硫酸钠,溶于水中,并稀释至100 mL。
酸性硫酸钠溶液:100 mL硫酸钠溶液(45 g/L)中,加2.5mL盐酸,混匀。
二硫化碳一四氯化碳混合液(1+200)。
盐酸(1+1):取50 mL盐酸,用水稀释至100 mL。
氢氧化钠溶液(240 g/L);取24 g氢氧化钠,加水溶解至100 mL。
三氯化铁溶液(50 g/L)。
硫酸铜溶液(35 g/L):称取3.5g硫酸铜(CuS·5O)溶于100 ml。水中。
酚酞一乙醇指示液(10 g/L),称取1 g酚酞,加乙醇(95%)溶解并稀释至100 mL。
马拉硫磷标准溶液:先将50 mL容量瓶准确称量,然后滴入约50 mg马拉硫磷,再准确称量,加四氯化碳至刻度,混匀,并计算其浓度。
马拉硫磷标准使用液:临用前于马拉硫磷标准溶液中加四氯化碳稀释至每毫升相当于100.0马拉硫磷。
仪器设备
分光光度计。
分析步骤称取约20. 00 g粉碎并全部通过20目筛的试样,置于200 mL具塞锥形瓶中,加40 mL四氯化碳,密塞,振荡2h,然后过滤。
吸取20 mL滤液于125 mL分液漏斗中,加0.2 mL二硫化碳一四氯化碳混合液,加10 mL酸性硫酸钠溶液,振摇1 mln静置分层,将四氯化碳层转入另一分液漏斗中,弃去水层。
吸取O、0.5、1.0、1.5、2.0、2.5 mL马拉硫磷标准使用液(相当O、50、100、150、200、250 马拉硫磷),分别置于125 mL分液漏斗中,加四氯化碳至20 mL,再各加0.2 mL二硫化碳一四氯化碳混合液。
于试样溶液及马拉硫磷标准溶液中各加5 mL无水乙醇,加0.4mL氢氧化钠溶液(240 g/L),准确激烈振摇1 min,立即加入10 mL硫酸钠溶液(45 g/L),混匀,加1滴酚酞指示液,用盐酸(1+1)中和至酚酞将褪色,再用盐酸(1+11)调pH为3~4(pH试纸试),再加0.5 mL三氯化铁溶液(50 g/L)。振摇1 min,静置分层(如乳化可离心分离),弃去四氯化碳层,用2 mE四氯化碳洗涤水层,振摇l min,分层后弃去四氯化碳层,如四氯化碳层带黄色,再用四氯化碳洗涤水层。在水层中准确加入4.0 mL四氯化碳、0.5 mL硫酸铜溶液(35 g/L),准确振摇1 min。静置分层后将四氯化碳层通过脱脂棉滤入2 cm比色杯中,以四氯化碳调节零点,在20 min内于波长415 nm处测吸光度,绘制标准曲线比较。
甲拌磷、杀螟硫磷、倍硫磷、敌敌畏、乐果、对硫磷
以气相色谱法按GB/T 5009. 20操作。
磷化物定性
原理
磷化物遇水和酸放出磷化氢,与硝酸银生成黑色磷化银,如有硫化物存在,同时放出硫化氢,与硝酸银生成黑色硫化银,干扰钡4定,而硫化氢又能与乙酸铅生成黑色硫化铅,以此证明是否有硫化物干扰。
试剂
酒石酸。
硝酸银溶液(100 g/L):贮存于棕色瓶中。
乙酸铅溶液(loo g/L)。
乙酸镉溶液(100 g/L)。
仪器
取2OO mL~250 mL锥形瓶,配一适宜双孔软木塞或橡皮塞,每孔内塞以内径0.4 cm~0.5 em、长 5 cm的玻璃管,每管内悬挂一长7 cm、宽0.3 cm~0.5 cm的滤纸条,临用时,一纸条用硝酸银溶液湿润,另一纸条用乙酸铅溶液湿润。
分析步骤
迅速称取2O. 00 g试样,置于锥形瓶中,加适量水至浸没试样,再加约0.5g酒石酸,立即塞好准备好的双孔塞,使滤纸条末端距液面约5 cm,在暗处置40~50水浴内加热30 min,观察试纸颜色变化情况。如试纸均不变色,表明磷化物负反应或未超过规定;如硝酸银试纸变色,乙酸铅试纸不变色,表示
可能有磷化物存在,需再定量;如两种纸均变色,可能有磷化物和硫化物同时存在或仅有硫化物存在,遇此情况,重取试样,加水后再加5 mL乙酸镉溶液(100 g/L),使形成黄色硫化镉沉淀,立即密塞,放置10 min,再加酒石酸,操作同前,如硝酸银试纸变黑,乙酸铅试纸不变色,表示有磷化物存在,需再定量。
定量
原理
磷化物遇水和酸,放出磷化氢,蒸出后吸收于酸性高锰酸钾溶液中被氧化成磷酸,与钼酸铵作用生成磷铝酸铵,遇氯化亚锡还原成蓝色化合物钼蓝,与标准系列比较定量。
本方法取样量为50 g时,检出限0.020 mg/kg。
试剂
高锰酸钾溶液(16.5 g/L):称取16.5g高锰酸钾,加水溶解后稀释至1 000 mL,静置三天或加热煮沸3 min,冷却,放置过夜,用玻璃棉或石棉过滤备用。
高锰酸钾溶液(3.3 g/L):将高锰酸钾溶液(16.5 g/L)用水稀释5倍。
硫酸(1十17):取28 mL硫酸缓缓加入400 mL水中,冷却后加水至500 mL。
硫酸(1+5):取83.3 mL硫酸缓缓加入400 mL水中,冷却后加水至500 mL。
饱和亚硫酸钠溶液:取28.5 g无水亚硫酸钠,加约70 mL水,微热溶勰后,放冷,稀释至100 mL。
钼酸钱溶液(50 g/L)。
氯化亚锡溶液;取0.lg氯化亚锡,溶于5 mL盐酸中,临用时现配。
磷化物标准溶液:准确称取0.040 0 g经105干燥过的无水磷酸二氢钾,溶于水,移入100 mL容量瓶中,加水稀释至刻度(可加一1滴三氯甲烷以增加保存时间),此溶液每毫升相当于0. 10 mg磷化氢。
磷化物标准使用液:吸取lO. OmL磷化物标准溶液,置于100 mL容量瓶中,加水至刻度。混匀-此溶液每毫升相当于10 磷化氢。
盐酸(1+1)。
饱和硝酸汞溶液。
饱和硫酸肼溶液。
酸性高锰酸钾溶液:高锰酸钾溶液(16.5 g/L)和硫酸(2 mol/L)等量混合。
仪器
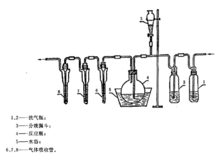
仪器如图所示。

分析步骤
试样测定;于三个串联的气体吸收管中各加5 mL高锰酸钾溶液(3.3 g/L)和1 mL硫酸(1+17)。二氧化碳发生瓶中装大理石碎块,从分液漏斗1中加适量的盐酸(1+1),作为二氧化碳发生器。二氧化碳气体顺序经装有饱和硝酸汞溶液、酸性高锰酸钾溶液、饱和硫酸肼溶液的洗气瓶洗涤后,进入反应瓶中(如用氮气代替二氧化碳,可以只通过硫酸肼溶液安全瓶直接进入反应瓶)。预先通二氧化碳(或氮气)5 min,打开反应瓶的塞子,迅速投入称好的509试样,立即塞好瓶塞,加大抽气速度使分液漏斗6中的5 mL硫酸(1+17)和80 mL水加至反应瓶中,然后减慢抽气和二氧化碳(或氮气)气流速度,将放置反应瓶的水浴加热至沸30 min,并继续通入二氧化碳(或氮气)。反应完毕后,先除去气体吸收管迸气的一端,再除去抽气管的一端,取下三个气体吸收管,分别滴加饱和亚硫酸钠溶液使高锰酸钾溶液褪色,合并吸收管中的溶液至50 mL比色管中,气体吸收管用少量水洗涤,洗液并人比色管中,加4.4mL硫酸(1+5),2.5 mL铝酸铵溶液(50 g/L),混匀。
吸取0、0.1、0.2、0.3、0.4、0.5mL磷化物标准使用液(相当于0、1、2、3、4、5 磷化氢),分别放人50 mL比色管中,加30 mL水,5.4 mL硫酸(1-1-5),2.5 mL钼酸铵溶液(50 g/L),混匀。予试样及标准管中各加水至50 mL混匀,再各加0.1 mL氯化亚锡溶液,混匀。1S rain后。用3 cm比色杯,以零管调节零点,于波长680 nm处测吸光度,绘制标准曲线比较l或与标准系列目测比较。取与处理试样量相同的试剂,按同一操作方法做试剂空白试验。
结果计算
试样中磷化物的含量按式(2)进行计算。
= (A1-A2)*1000/(m*1000)……………………………………(2)
式中:
——试样中磷化物的含量(以磷化氢计),单位为毫克每千克(mg/kg);
——测定用试样磷化物的质量,单位为微克();
——试剂空白中磷化物的质量,单位为微克();
——试样质量,单位为克(g)。
计算结果保留两位有效数字。
以空气代替二氧化碳-空气顺序经装有酸性高锰酸钾溶液、碱性焦性没食子酸溶液(5 g焦性没食子酸溶于15 mL水,48 g氢氧化钾溶于32 mL水中,然后两液混合)的洗气瓶洗涤后进入反应瓶。以下操作同上。其装置如图所示。
氰化物定性
原理
氰化物遇酸产生氢氰酸,氢氰酸与苦味酸钠作用,生成红色异氰紫酸钠。
试剂
洒石酸。
碳酸钠溶液(100 g/L)。
苦味酸试纸:取定性滤纸剪成长7 cm、宽0.3 cm~O.5 cm的纸条,浸入饱和苦昧酸-乙醇溶液中,数分钟后取出,在空气中阴干,贮存备用。
仪器
取200 mL~300 mI,锥形瓶,配备一适宜的单孔软木塞或橡皮塞,孔内塞以内径0.4 cm~0.5 cm,长5 cm的玻璃管,管内悬一条苦味酸试纸,临用时,试纸条以碳酸钠溶液(100 g/L)湿润。
分析步骤
迅速称取5 g试样,置于100 mL锥形瓶中,加20 mL水及0.5 g酒石酸,立即塞上悬有苦味酸并以
碳酸钠湿润的试纸条的木塞,置40~50水浴中,加热30min,观察试纸颜色变化。如试纸不变色,表示氰化物为负反应或未超过规定l如试纸变色,需再做定量试验。
定母
原理
氰化物在酸性溶液中蒸出后被吸收于碱性溶液中,在pH 7.0溶液中,用氯胺T将氰化物转变为氯化氰,再与异烟酸一吡唑酮作用,生成蓝色染料,与标准系列比较定量。
本方法取样量为10 g时,检出限为0. Ol5mg/kg。
试剂
甲基橙指示液(0.5 g/L)。
乙酸锌溶液(100 g/L)。
酒石酸。
氢氧化钠溶液(10 g/L)。
氢氧化钠溶液(1 g/L)。
乙酸(1十24)。
酚酞一乙醇指示液(10 g/L)。
磷酸盐缓冲溶液[(0.5 mol/L) pH7.O]:称取34.0g元水磷酸二氢钾和35.5 g无水磷酸氢二钠,溶于水并稀释至1 000 mL。
试银灵(对二甲氨基亚苄基罗丹宁)溶液:称取0.02 g试银灵,溶于100 mL丙酮中。
异烟酸一吡唑酮溶液:称取1.5g异烟酸溶于24 mL氢氧化钠溶液(20 g/L)中,加水至100 mL,另称取0.25g吡唑酮,溶于20 mLN-二甲基甲酰胺中,合并上述两种溶液,混匀。
氯胺T溶液:称取1 g氯胺T(有效氯含量应在11%以上),溶于100 mL水中,临用时现配。
氰化钾标准溶液:称取0.25 g氰化钾,溶于水中,并稀释至1 000 mL,此溶液每毫升约相当于0.1 mg氰化物,其准确度可在使用前用下法标定。
取上述溶液10.O mL,置于锥形瓶中,加1 mL氢氧化钠溶液(20 g/L),使pH为11以上,加0.1 mL试银灵溶液,用硝酸银标准溶液(0. 020 mol/L)滴定至橙红色[1 mL硝酸银标准溶液(0. 020 mol/L)相当于1.08 mg氢氰酸]。
氰化钾标准使用液;根据氰化钾标准溶液的浓度吸取适量,用氢氧化钠溶液(1g/L)稀释成每毫升相当于1 氢氰酸。
仪器
250mL玻璃水蒸气蒸馏装置。
分光光度计。
分析步骤
迅速称取10.00 g试样,放置于250 mL蒸馏瓶中,加适量水使试样全部浸没,加20 mL乙酸锌溶液(100 g/L),加1 g~2 g酒石酸,迅速连接好全部装置,冷凝管下端插入盛有5 mL氢氧化钠溶液(10 g/L)的100 mL容量瓶的液面下,缓缓加热,通水蒸气进行蒸馏,收集馏液近100 mL,取下容量瓶,加水至刻度,混匀,取10 mL蒸馏液置于25 mL比色管中。
吸取O、0.3、0.6、0.9、1.2、1.5 mL氰化物标准溶液(相当于0、0.3、0.6、0.9、1.2、1.5 #g氢氰酸),分别置于25 mL比色管中,各加水至10 mL。于试样溶液及标准溶液中各加1 mL氢氧化钠溶液(10 g/L)和1滴酚酞指示液,用乙酸(1+24)调至红色刚剐消失,加5 mL磷酸盐缓冲溶液,加温至37左右,再加入0.25 mL氯胺T溶液,加塞混合,放段5 min,然后加入5 mL异烟酸一吡唑酮溶液,加水至25 mL,混匀,于25℃"-,40℃放置40 min,用2 cm比色杯,以零管调节零点,予波长638 nm处测吸光度,绘制标准曲线比较。
结果计算
试样中氰化物(以氢氰酸计)的含量按式(3)进行计算。
X= A*1000/(m*V1/v2*1000) ……………………………………(3)
式中:
——试样中氰化物(以氢氰酸计)的含量,单位为毫克每千克(mg/kg);
——测定用试样液氢氰酸的质量,单位为微克();
——试样质量,单位为克(g);
——试样蒸馏液总体积,单位为毫升(mL);
——测定用蒸馏液体积,单位为毫升(mL)。
计算结果保留两位有效数字。
氯化苦原理
氯化苦可被乙醇钠分解形成亚硝酸盐。在弱酸性溶液中与氨基苯磺酸进行重氮化,然后再与N-l-萘基乙二胺盐酸偶合生成紫红色,与标准系列比较定量。
本方法取样量20 g时,检出限为0.050 mg/kg。
试剂
乙醇钠溶液:取金属钠,先用滤纸将表面煤油吸干,并用小刀切去表面被氧化部分(切下表面部分,务必放回煤油中,切勿与水接触),然后取5 g切成碎片,量取1 000 mL无水乙醇,置于大烧杯中,将切好的金属钠立即分次加入,待作用完毕,杯中不再有气体发生时,移人棕色瓶中备用。
无水乙醇。
对氨基苯磺酸溶液(4 g/L):称取0.4g对氨基苯磺酸溶于lOOmL盐酸(1+1)中,贮存于棕色瓶中。
N-1-萘基乙二胺溶液(2 g/L)l称取0.2 9 N-l-萘基乙二胺,溶于100m水中,贮存于棕色瓶中。
氯化苦标准储备溶液:量取约20 mL无水乙醇,置于50 mL容量瓶中,准确称量后,加入2滴氯化苦,再准确称量,两次的差即为氯化苦质量,加无水乙醇至刻度,混匀。
氯化苦标准使用液:吸取适量氯化苦标准储备溶液,置于50 mL容量瓶中,加无水乙醇稀释至刻度,此溶液每毫升应相当于0. 020 氯化苦,贮存于冰箱中。
仪器
分光光度计。
分析步骤
称取约20.00 g试样,置于100mL具塞锥形瓶中,加20 mL乙醇钠溶液,盖好,放置暗处8 h~10 h或过夜,过滤,量取5.O mL滤液,置于10 mL比色管中。
吸取5.0mL氯化苦标准使用液a置于50 mL容量瓶中,加20 mL乙醇钠溶液,放置暗处8 h~10 h或过夜,再加无水乙醇稀释至刻度,然后吸取0、1.0、2.0、3.0、4.O、5.0 mL此液(相当于O、2、4、6、8、10 氯化苦),分别置于I0 mL比色管中,再各加元水乙醇至5 mL,加乙酸(36%)1 mL。
于试样及标准管中各加1 mI。对氨基苯磺酸(4 g/L),混匀,静置3 min~5 min后,各加入0.5 mLN-I-萘基乙二胺溶液(2 g/L)。加无水乙醇至刻度,混匀后放置20 min,用1 cm比色杯,以零管调节零点,于波长538 nm处测吸光度,绘制标准曲线比较。
4.5.5结果计算
试样中氯化苦的含量按式(4)进行计算。
X= A*1000/(m*V1/v2*1000)……………………………………(4)
式中:
——试样中氯化苦的含量,单位为毫克每千克(mg/kg);
——测定用试样中氯化苦的质量,单位为微克();
——试样中加入乙醇钠总体积。单位为毫升(mL);
——测定用试样滤液的体积,单位为毫升(mL);
——试样质量,单位为克(g)。
计算结果保留两位有效数字。
二硫化磷原理
二硫化碳与二乙胺作用生成二乙胺磺酸,再与铜盐反应生成黄色复盐,与标准系列比较定量。
本方法取样量为25 g时,检出限为0. 20 mg/kg。
试剂
二乙胺-乙醇溶液(10 g/L)。
乙酸铜-乙醇溶液(0.5 g/L)。
硫酸铜溶液(50 g/L)。
二硫化碳标准储备溶液:量取约20mL二乙胺一乙醇溶液(10 g/L),置于50 mL容量瓶中,准确称量后,加入2滴二硫化碳,再准确称量,两次的差即为二硫化碳质量,加二乙胺一乙醇溶液(10 g/L)至刻度,混匀。
硫化碳标准使用液:吸取适量二硫化碳标准溶液,置于50 mL容量瓶中,加二乙胺一乙醇溶液(lO g/L)稀释至刻度,此溶液每毫升相当于0.020 mg=硫化碳。
乙醇(95%)。
仪器
分光光度计。
分析步骤
称取约26. 00 g试样,置于600 mL圆底烧瓶中,加入水适量至浸没试样,于气体吸收管内各加入110 mL二乙胺-乙醇溶液(10 g/L),洗气瓶中加入50mL硫酸铜溶液(50 g/L),以除去空气中的硫化物,三将圆底烧瓶置于70℃左右的水浴锅中,按图3连接好洗气瓶、吸收管、抽气装置,缓缓抽气2h后,取下气体吸收管。停止加热和抽气。将气体吸收管中吸收液倒入50 mL容量瓶中,用少量二乙胺一乙醇溶液(10 g/L)洗气体吸收管
2次~3次,洗液并入容量瓶中,再用二乙胺一乙醇溶液(10 g/L)加至刻度,混匀。吸取5.0 mL置于125 mL比色管中。
吸取O、0.5、1.0、1.6、2.0、2.5mL一二硫化碳标准使用液(相当0、10、20、30、40、50二硫化碳),分别置于25 mL比色管中,再各加二乙胺·乙醇溶液(10 g/L)至10 mL,混匀。
于试样及标准管中各加1 mL乙酸铜一乙醇溶液(0.5 g/L),再加乙醇至刻度,混匀。用1 cm比色杯,以零管调节零点,于波长400 nm处测吸光度,绘制标准曲线比较。
4.6.5结果计算
试样中二硫化碳的含量按式(5)进行计算。
X= A*1000/(m*V1/v2*1000) ……………………………………(5)
式中:
——试样中氯化苦的含量,单位为毫克每千克(mg/kg);
——测定用试样中氯化苦的质量,单位为微克();
——试样中加入乙醇钠总体积。单位为毫升(mL);
——测定用试样滤液的体积,单位为毫升(mL);
——试样质量,单位为克(g)。
计算结果保留两位有效数字。
砷
按GB/T 5009. 11操作。
汞
按GB/T5009. 17操作。
六六六、滴滴涕
按GB/T 5009.19操作。
黄曲霉薅素
按GB/T 5009.22操作。
镉
按GB/T 5009.15操作。
氟
按GB/T 5009.18操作。
曼陀罗籽
鉴别
曼陀罗籽星三角形或肾形,扁平,表面有网点,呈棕色或棕褐色,有的边缘有皱折,淡棕色,宽2 mm~3 mm,有的较大,约5 mm~6 mm。
生物碱比色定性原理
试样中所含阿托品等生物碱经提取后与发烟硝酸及氢氧化钾溶液有呈色反应。
试剂
氨水(1+1)。
乙醚。
盐酸(1十5)。
三氯甲烷。
无水硫酸钠。
发烟硝酸。
氢氧化钾-乙醇溶液(100 g/L)。
分析步骤
将约30粒曼陀罗籽故人乳钵中,加氨水(1十1)浸湿,浸渍片刻,研磨成粘稠状,加乙醚研磨三次,每次10 ml,,将乙醚合并于分液漏斗中,加10 mL盐酸(1+5),振摇提取l min,分出盐酸层至另一分液漏斗中,加氨水(1+1)调成碱性,用10 mL三氯甲烷振摇提取1 min,再提一次,合并三氯甲烷层,通过无水硫酸钠脱水后浓缩至0.5 mL,备用。
取0.2 mL试液于小蒸发皿中,挥干溶剂,加4滴发烟硝酸使残渣溶解,水浴上蒸于,残留物变黄色,冷却后加数滴氢氧化钾一乙醇溶液(300 g/L),则变紫堇色,随即变红色。阿托品、莨菪碱和东莨菪碱均有此反应。
薄层色谱定性原理
试样中所含阿托品等生物碱经提取后,用薄层分离,再以显色剂显色,与对照标准比较。
试剂
硅胶G薄层板:厚度0.3 mm~O. 5mm,105℃活化lh,放干燥器中备用。
展开剂;甲醇-氨水(200十3)。
显色剂:称取0.85 g次硝酸铋,加10 mL,冰乙酸,加40 mL水,溶解。取5 mL,加5 mL碘化钾溶液(4 g碘化钾溶于5 mL水中),再加20 mL冰乙酸,加水稀释至100 mL。
阿托品标准溶液:称取120.0 mg硫酸阿托品,溶于10 mL水中,加氨水(1十1)显碱性,用三氯甲烷提取二次,每次8 mL,三氯甲烷提取液经少许无水硫酸钠脱水,滤人20 mL具塞比色管中,再用少许三氯甲烷洗滤器,洗液并入比色管中,加三氯甲烷至20 mL,此溶液每毫升相当于5.O mg阿托品。
东莨菪碱标准溶液:称取145.O mg氢溴酸东莨菪碱,以下按阿托品标准溶液同样处理,配成每毫升相当于5.0 mg东莨菪碱。
分析步骤
在薄层板下端2 cm处,点10阿托品及东茛菪碱标准溶液,30 ~lOO试样提取浓缩液,各点间距1.5 cm,置于预先用展开剂饱和的展开槽中,待溶剂前沿上展至I0 cm~15 cm,取出,挥干展开剂,喷显色剂呈现橙红色斑点为阳性反应。
定置
称取1 000 g粮食,从中检出曼陀罗籽,不得超过5粒。
麦角鉴别
形态
麦角呈三条或四条钝的圆柱形,微弯:两端稍窄细,长0.3 cm~O.4 cm。粗1 mm~7 mm,外面呈黑色或紫棕色,有纵沟与横裂纹,质脆,易折断,断面钝三角形,边缘暗紫色,中心呈灰白或紫白色。
组织切片
将麦角泡入水中,浸泡24 h,使膨腑夹在土豆或萝卜中间并固定,用小手术刀切成尽可能薄的小片,用次甲基蓝溶液(1 g/L)显色,在显微镜下观察,其组织紧密。
麦角红素和麦角生物碱定性
试剂
酒石酸溶液(20 g/L)。
无水乙醚。
饱和碳酸氢钠溶液。
氨水(1十1)。
三氯甲烷。
对二甲氨基苯甲醛溶液。称取0.125 g对二甲氨基举甲醛,加100 mL稀硫酸(65 mL硫酸缓缓倒入35 mL水中,混匀,冷却)溶解,然后加0.1mL三氯化铁溶液(50 g/L),混匀。
硫酸。
无水乙醇:紫外光灯波长365 nm下观察无荧光。
乙酸乙酯。
过氧化氢溶液(3%)。
分析步骤
取20粒可疑麦角予研钵中,研碎,加酒石酸溶液(20 g/L)研成粘稠状,加10 mL乙醚,仔细研磨,分取乙醚,反复进行三次,合并乙醚层于试管中,保留残渣于研钵内。在试管内加0.5 mL饱和碳酸氢钠溶液,振摇,碳酸氢钠溶液层变红色,即表示检出麦角红。
取保留的残渣,加氨水(1+1)研磨呈碱性,用三氯甲烷提取三次,每次10 mL,合并三氯甲烷层,分成两部分,一部分小心加2 mL对二甲氨基苯甲醛溶液,在两液层接触面呈蓝紫色环,数分钟后,三氯甲烷层均显蓝色,即表示检出麦角生物碱。另一部分三氯甲烷提取液置于试管中,在热水浴上使三氯甲烷尽,残留物加无水乙醇溶解,在波长365 nm紫外光灯下观察有强烈蓝色荧光,即表示检出麦角生物碱。
定量
称l 000 g粮食,检出麦角后称量,不得超过0.lO g。
毒麦根据形态鉴别,毒麦籽实被内外稃紧包。紧贴于内稃内,其芒联接于外稃部,芒长7 mm~15 mm,籽实长椭圆型,坚硬无光泽,呈灰褐色,长4 mm~6 mm,腹沟较宽,籽实1 000粒,质量10g~13 g,
定量;称取1 000 g粮食,拣出的毒麦不得超过1 g。
二溴乙烷
蒸馏法(SGS法)
试剂
己烷:重蒸,使用前以气相色谱法检验,应无干扰蜂。
去泡剂(baker andifoam B)。
无水硫酸钠。
二溴乙烷(EDB)标准使用液:用己烷配制成含1,0、2.5、5.0、10.0 ng/mL EDB的标准使用液。
仪器
气相色谱仪:附有电子捕获检测器。
分析步骤
试样处理
快速称取50.0 g试样(贮存子5℃以下)于1 000 mL圆底烧瓶中,加300 mL水、10.0 mL己烷,连接烧瓶、接收管、冷凝管,放在加热器上缓缓加热,用去泡剂避免过多的发泡[几滴硅油或液体石蜡或吐温(Tween) 80],加热到已烷全部蒸出,水分开始在已烷层下集聚,移去加热器,待其冷却后,记下回收的己烷毫升数,并转入具塞试管内,加2 9~3 9无水硫酸钠脱水,供气相色谱测定。
气榻色谱参考条件
色谱柱;2 m×3 mm(内径)不锈钢柱,内装涂有10~% Squalene的80目~100目Chromosorb W HP。
温度:柱温:75℃;检测器,275℃或300℃;进样口:200℃;
载气:氮气50 mL/min。
测定
准确吸取2 #L样液及不同浓度EDB标准使用液,注入气相色谱仪中,每个试样重复三次,取平均峰高,以标准浓度值对相应的色谱峰高值作EDB标准曲线,再以样液峰高与标准曲线比较定量。
4.16.1.3.4结果计算
试样中二溴乙烷的含量按式(6)进行计算。
X= A*1000/(m*V1/v2*1000) ……………………………………(6)
式中:
——试样中二溴乙烷的含量,单位为微克每千克(mg/kg);
——进样样液相当二溴乙烷的质量,单位为纳克();
——试样进样体积,单位为微升(mL);
——收集的己烷蒸馏液的体积,单位为毫升(mL);
——试样质量,单位为克(g)。
计算结果保留两位有效数字。
浸渍法
试剂
丙酮:重蒸,使用前以气相色谱法检验,应无干扰峰。
无水氯化钙。
氯化钠。
丙酮-水(5+1)。
二溴乙烷标准使用液:用丙酮配制并稀释至每毫升含0.2 二溴乙烷。
仪器
气相色谱仪,附有电子捕获检测器。
分析步骤
试样处理
快速称取50.0 g试样(贮存于5℃以下),置于250mL具塞锥形瓶中,加150mL丙酮一水溶液,密塞,摇匀,在20℃~25℃暗处浸泡48 h。24 h振摇一次,吸取10.0 mL上清液于25 mL具塞试管中,加2 g氯化钠,密塞,剧烈振摇2 min,放置分层后,吸取5.O mL上清液于10 mL具塞试管中,加1 g尼水氯化钙,密塞,剧烈振摇2 min,静置30 min以上,供气相色谱测定用。
气相色谱参考条件
色谱柱=2m×3 mm(内径)不锈钢柱,内装涂有15%聚丙二醇(Poly propylene glycol)或15%Ucon LB-550X的60目~80目Chromosorb W。
温度:柱温:140℃;检测器、进样口温度,200℃。
载气:氮气70 mL/min。
测定
每天做标准曲线。吸取O、0.5、1.0、3.O、5.O、7.O 二溴乙烷标准使用液(相当予O、0.1、0.2、0.6、1.O、1.4 ngEDB),直接进样,与测得峰高绘制标准曲线。
准确吸取0.5 L试样澄清液,进样。为避免检测器超载,可将样液用无水丙酮稀释10倍或100倍再进样。每个样液都进样3次,取平均峰高与标准曲线比较定量。
结果计算
试样中二溴乙烷的含量按式(7)进行计算。
X= A*1000/(m*V1/v2*1000) ……………………………………(6)
式中:
——试样中二溴乙烷的含量,单位为微克每千克(mg/kg);
——进样样液相当二溴乙烷的质量,单位为纳克();
——试样进样体积,单位为微升(mL);
——试样质量,单位为克(g)。
125——150mL浸渍液中丙酮的体积,单位为毫升(mL)。
注:如无以上介绍的固定液,以下色谱柱也可用。
①20%OV-101涂于Chromosorb W HP 80目~100目上。
②30% DC-200涂于Gas Chrom Q 80目~100目上。
③10%DC-200涂予Chromosorb W AW DMCS 60目~80目上。
④6%OV-210+F4% SE-30涂于Gas Chrom Q 80目~100目上。
计算结果保留两位有效数字。
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国