

化学反应是指分子破裂成原子,原子重新排列组合生成新分子的过程,称为化学反应。在反应中常伴有发光发热变色生成沉淀物等,判断一个反应是否为化学反应的依据是反应是否生成新的分子。12
核反应不属于化学反应。
实质化学反应的本质是旧化学键断裂和新化学键形成的过程。
在反应中常伴有发光、发热、变色、生成沉淀物等。判断一个反应是否为化学反应的依据是反应是否生成新的物质。根据化学键理论,又可根据一个变化过程中是否有旧键的断裂和新键的生成来判断其是否为化学反应。1
化学反应类型按反应物与生成物的类型分四类:化合反应、分解反应、置换反应、复分解反应
按电子得失可分为:氧化还原反应、非氧化还原反应;氧化还原反应包括:自身氧化还原,还原剂与氧化剂反应
异构化:(A →B) :化合物是形成结构重组而不改变化学组成物。1
化学合成:化合反应
简记为:A + B = C:二种以上元素或化合物合成一个复杂产物。(即由两种或两种以上的物质生成一种新物质的反应。)
化学分解:分解反应
简记为:A = B + C :化合物分解为构成元素或小分子。(即化合反应的逆反应。它是指一种化合物在特定条件下分解成两种或两种以上较简单的单质或化合物的反应。)1
置换反应(单取代反应)
简记为:A+BC=B+AC :表示额外的反应元素取代化合物中的一个元素。(即指一种单质和一种化合物生成另一种单质和另一种化合物的反应。)1
(置换关系是指组成化合物的某种元素被组成单质的元素所替代。置换反应必为氧化还原反应,但氧化还原反应不一定为置换反应。)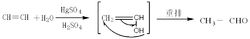 根据反应物和生成物中单质的类别,置换反应有以下4种情况:
根据反应物和生成物中单质的类别,置换反应有以下4种情况:
①较活泼的金属置换出较不活泼的金属或氢气
②较活泼的非金属置换出较不活泼的非金属
③非金属置换出金属
④金属置换出非金属
(详细请见置换反应词条……)3
复分解反应(双取代反应)
简记为:AB+CD=AD+CB :在水溶液中(又称离子化的)两个化合物交换元素或离子形成不同的化合物。(即由两种化合物互相交换成分,生成另外两种化合物的反应。)
复分解反应的本质是溶液中的离子结合成难电离的物质(如水)、难溶的物质或挥发性气体,而使复分解反应趋于完成。酸、碱、盐溶液间发生的反应一般是两种化合物相互交换成分而形成的,即参加反应的化合物在水溶液中发生电离离解成自由移动的离子,离子间重新组合成新的化合物,因此酸、碱、盐溶液间的反应一般是复分解反应。复分解反应是离子或者离子团的重新组合,因为此类反应前后各元素的化合价都没有变化,所以复分解反应都不是氧化还原反应。
当然还有更多复杂的情形,但仍可逐步简单化而视为上述反应类别的连续反应。化学反应的变化多端难以建立简单的分类标准。 但是一些类似的化学反应仍然可以归类,譬如:
歧化反应:
指的是同一物质的分子中同一价态的同一元素间发生的氧化还原反应。同一价态的元素在发生氧化还原反应过程中发生了“化合价变化上的分歧”,有些升高,有些降低。发生歧化反应的元素必须具有相应的高价态和低价态化合物,歧化反应只发生在中间价态的元素上。氟(F2)无歧化作用,因为氟元素电负性最大,无正化合价,只有负化合价。
归中反应与歧化反应均属同种物质间发生的氧化还原反应,歧化反应是自身氧化还原反应的一种,但自身氧化还原反应却不一定都是歧化反应。
归中反应(反歧化反应):
指的是物质中不同价态的同种元素之间发生的氧化还原反应。即同一元素的价态由反应前的高价和低价都转化成反应以后的中间价态,在化学反应中元素的价态变化有个规律:只靠拢,不交叉。因此元素的高价和低价都只能向中间靠拢。归中反应和歧化反应是两个‘相反’的过程,这两种反应都一定是氧化还原反应。
有机反应:指以碳原子化合物为主的各种反应。
氧化还原反应:指两化合物间的电子转移(如:单取代反应和燃烧反应)
燃烧反应(初中化学书上也叫氧化反应):指物质和氧气的反应。
氯化反应;指氯与有机物反应;
更多的例子参见化学反应列表(list of reactions)。
|| ||
核反应不是化学反应,核反应属于核物理变化。原子是化学变化中最小化的粒子,化学反应我们也可以看作是原子的重新组合,在反应前后元素种类和原子个数是保持不变的。而核反应中的原子变成了其他的原子了,故不是化学变化。
反应能量能量净改变
根据热力学第二定律,任何等温等压封闭系统倾向降低吉布斯自由能。在没有外力的影响下,任何反应混合物也是如此。比方,对系统中焓的分析可以得到合乎反应混合物的热力学计算。反应中焓的计算方式采用标准反应焓以及反应热加成性定律(赫士定律)。
以一个甲烷和氧气的燃烧反应为例:
CH4 + 2O2 →CO2 + 2 H2O
能量计算须打断反应左侧和右侧的所有键结取得能量数据,才能计算反应物和生成物的能量差。以 ΔH 表示能量差。Δ(Delta) 表示差异,H 则为焓等于固定压力下的热传导能量。ΔH 的单位为千焦耳或千卡。
反应判断假设,存在一个系统。通过计算得出反应前后系统的焓变为ΔH。
如果ΔH 计算为负值,则反应必为放热反应。比如:燃烧就是燃料与空气中氧气剧烈反应放出热量。
如果ΔH 计算是正值,则反应必为吸热反应。比如:石灰石在高温下反应分解出氧化钙(生石灰)和二氧化碳。
如果ΔH计算等于零,则反应不吸热也不放热;外界不对体系做功,体系也不对外界做功。
反应到底是吸热还是放热完全由体系的 ΔH决定。但是,反应是否进行则完全由体系的由吉布斯自由能ΔG来表示。具体计算公式如下:
ΔG = ΔH - TΔS1
ΔG 是自由能改变,ΔH 是焓改变,而ΔS 则为熵改变,T是开尔文温度。
反应中间物当热力学企图回答这个问题:“反应会发生吗?”,另一个重要问题“反应多快?”却完全没有回答。这是因为热力学或者热力学平衡试著要了解的是反应混合物初始和结束状态。因此无法指出反应发生时的过程。这个领域属于反应动力学的范畴。
基元反应是如何发生的?传统观点认为,反应物碰撞形成所谓活化复合物。碰撞的动能使活化复合物获得更高的能量,导致构成反应的键结重组。但是,这种观点导致了一个困境:活化复合物的结构和能量不能同时确定,否则有悖于测不准原理。所以,一些物理学家认为,实际上这些复合物未必真的存在,而是能量空间的一些隔离面。相对的活化络合物的观点更多的是为实验化学家所接受,因为在描述反应机理时比较方便。现代理论化学已经可以精确的计算速率常数。对于一些比较简单的反应,整个过程的态分辨信息也可以获得。
反应条件指化学反应所必须或可提高反应速率的方法,如:加热(△)、点燃、高温、电解、通电(电解)、紫外线或催化剂等。
反应速率化学反应的反应速率是相关受质浓度随时间改变的的测量。反应速率的分析有许多重要应用,像是化学工程学或化学平衡研究。反应速率受到下列因素的影响:
反应物浓度:如果增加通常将使反应加速。
活化能:定义为反应启始或自然发生所需的最低能量。
愈高的活化能表示反应愈难以启始,反应速率也因此愈慢。
反应温度:温度提升将加速反应,因为愈高的温度表示有愈多的能量,使反应容易发生。
催化剂:催化剂是一种通过改变活化能来改变反应速率的物质。而且催化剂在反应过程中不会破坏或改变,所以可以重复作用。
反应速率与参与反应的物质浓度有关。物质浓度则可透过质量作用定律定量。
化学平衡根据吉布斯自由能判据,当ΔrGm=0时,反应达最大限度,处于平衡状态。化学平衡的建立是以可逆反应为前提的。可逆反应是指在同一条件下既能正向进行又能逆向进行的反应。绝大多数化学反应都具有可逆性,都可在不同程度上达到平衡。
从动力学角度看,反应开始时,反应物浓度较大,产物浓度较小,所以正反应速率大于逆反应速率。随着反应的进行,反应物浓度不断减小,产物浓度不断增大,所以正反应速率不断减小,逆反应速率不断增大。当正、逆反应速率相等时,系统中各物质的浓度不再发生变化,反应达到了平衡。
19世纪50-60年代,热力学的基本规律已明确起来,但是一些热力学概念还比较模糊,数字处理很烦琐,不能用来解决稍微复杂一点的问题,例如化学反应的方向问题。当时,大多数化学家正致力于有机化学的研究,也有一些人试图解决化学反应的方向问题。这种努力除了质量作用定律之外,还有其他一些人试图从别的角度进行反应方向的探索,其中已有人提出了一些经验性的规律。
化学变化的研究在这一时期,丹麦人汤姆生和贝特罗试图从化学反应的热效应来解释化学反应的方向性。他们认为,反应热是反应物化学亲合力的量度,每个简单或复杂的纯化学性的作用,都伴随着热量的产生。贝特罗更为明确地阐述了与这相同的观点,并称之为“最大功原理”,他认为任何一种无外部能量影响的纯化学变化,向着产生释放出最大能量的物质的方向进行。虽然这时他发现了一些吸热反应也可以自发地进行,但他却主观地假定其中伴有放热的物理过程。这一错误的论断在30年代终于被他承认了,这时他才将“最大功原理”的应用范围限制在固体间的反应上,并提出了实际上是“自由焓”的化学热的概念。 19世纪60-80年代,霍斯特曼、勒夏特列和范霍夫在这一方面也做了一定的贡献。首先,霍斯特曼在研究氯化铵的升华过程中发现,在热分解反应中,其分解压力和温度有一定的关系,符合克劳胥斯·克拉佩隆方程:dp/dt=Q/T(V'-V)其中Q代表分解热,V、V'代表分解前后的总体积。范霍夫依据一述方程式导出的下式:
19世纪60-80年代,霍斯特曼、勒夏特列和范霍夫在这一方面也做了一定的贡献。首先,霍斯特曼在研究氯化铵的升华过程中发现,在热分解反应中,其分解压力和温度有一定的关系,符合克劳胥斯·克拉佩隆方程:dp/dt=Q/T(V'-V)其中Q代表分解热,V、V'代表分解前后的总体积。范霍夫依据一述方程式导出的下式:
lnK=-(Q/RT) c
此式可应用于任何反应过程,其中Q代表体系的吸收的热(即升华热)。范霍夫称上式为动态平衡原理,并对它加以解释,他说,在物质的两种不同状态之间的任何平衡,因温度下降,向着产生热量的两个体系的平衡方向移动。1874年和1879年,穆迪埃和罗宾也分别提出了这样的原理。穆迪埃提出,压力的增加,有利于体积相应减少的反应发生。在这之后,勒夏特列又进一步普遍地阐释了这一原理。他说,处于化学平衡中的任何体系,由于平衡中的多个因素中的一个因素的变动,在一个方向上会导致一种转化,如果这种转化是惟一的,那么将会引起一种和该因素变动符号相反的变化。
然而,在这一方面做出突出贡献的是吉布斯,他在热力化学发展史上的地位极其重要。吉布斯在势力化学上的贡献可以归纳4个方面。第一,在克劳胥斯等人建立的第二定律的基础上,吉布斯引出了平衡的判断依据,并将熵的判断依据正确地限制在孤立体系的范围内。使一般实际问题有了进行普遍处理的可能。第二,用内能、熵、体积代替温度、压力、体积作为变量对体系状态进行描述。并指出汤姆生用温度、压力和体积对体系体状态进行描述是不完全的。他倡导了当时的科学家们不熟悉的状态方程,并且在内能、熵和体积的三维坐标图中,给出了完全描述体系全部热力学性质的曲面。第三,吉布斯在热力学中引入了“浓度”这一变量,并将明确了成分的浓度对内能的导数定义为“热力学势”。
这样,就使热力学可用于处理多组分的多相体系,化学平衡的问题也就有了处理的条件。第四,他进一步讨论了体系在电、磁和表面的影响下的平衡问题。并且,他导出了被认是热力学中最简单、最本质也是最抽象的热力学关系,即相律,在,而平衡状态就是相律所表明的自由度为零的那种状态。
吉布斯对平衡的研究成果主要发表在他的三篇文章之中。1873年,他先后将前两篇发表在康涅狄格州学院的学报上,立即引起了麦克斯韦的注意。吉布斯前两篇文可以说只是一个准备,1876年和1878年分两部分发表了第三篇文章《关于复相物质的平衡》,文章长达300多页,包括700多个公式。两篇文章是讨论单一的化学物质体系,这篇文章则对多组分复相体系进行了讨论。由于热力学势的引入,只要将单组分体系状态方程稍加变化,便可以对多组分体系的问题进行处理了。
对于吉布斯的工作,勒夏特列认为这是一个新领域的开辟,其重要性可以与质量不灭定律相提并论。然而,吉布斯的三篇文章发表之后,其重大意义并未被多数科学家们所认识到,直到1891年才被奥斯特瓦德译成德文,1899年勒夏特列译成法文出版之后,情况顿然改变。在吉布斯之后,热力学仍然只能处理理想状态的体系。这时,美国人洛易斯分别于1901年和1907年发表文章,提出了“逸度”与“活度”的概念。路易斯谈到“逃逸趋势”这一概念,指出一些热力学量,如温度、压力、浓度、热力学势等都是逃逸趋势量度的标度。
路易斯所提出的逸度与活度的概念,使吉布斯的理论得到了有益的补充和发展,从而使人们有可能将理想体系的偏差进行统一,使实际体系在形式上具有了与理想体系完全相同的热力学关系式。综上所述,化学平衡状态是指在一定条件下的可逆反应,正反应和逆反应的速率相等,反应混合物中各组分的浓度保持不变的状态。
化学平衡状态具有逆,等,动,定,变等特征。
逆:化学平衡研究的对象是可逆反应。
等:平衡时,正逆反应速率相等,即v正=v逆。
动:平衡时,反应仍在进行,是动态平衡,反应进行到了最大程度。
定:达平衡状态时,反应混合物中各组分的浓度保持不变,反应速率保持不变,反应物的转化率保持不变,各组分的含量保持不变。
变:化学平衡跟所有的动态平衡一样,是有条件的,暂时的,相对的,当条件发生变化时,平衡状态就会被破坏,由平衡变为不平衡,再在新的条件下建立新平衡。 影响化学平衡的因素有很多:如压强、温度、浓度等。
影响化学平衡的因素有很多:如压强、温度、浓度等。
注意:催化剂不影响化学平衡,仅影响反应速率。在其他条件不变的情况下,增大反应物浓度或减小生成物浓度,可使平衡向正反应方向移动。
勒夏特列原理:如果改变影响平衡的一个条件(浓度压强或温度等),平衡就向能够减弱这种改变的方向移动。
反应现象放热,吸热,发光,变色,产生沉淀,生成气体
可逆与自发反应每个化学反应理论上均是可逆反应。正反应中定义物质从反应物转换成产物。逆反应则相反,产物转换成反应物。
化学平衡指正反应速率和逆反应速率达到相等的状态,因此反应物和产物均会存在。然而,平衡态的反应方向可透过改变反应状态改变,譬如温度或压力。勒夏特列原理在此用来预测是产物或反应物形成。
虽然所有的反应在一些范围内均是可逆的,部份反应仍可归类为不可逆反应。“不可逆反应”指得是“完全反应”。意思是几乎所有的反应物均形成产物,甚至在极端状况下均难以逆转反应。
另一种反应机制称为自发反应,是一种热力学倾向,表示此反应引起总体熵的净增加。自发反应(相对于非自发反应)不须外在协助(如能量供给)就会产生。在化学平衡的系统中,反应过程中自发反应的方向可预期形成较多的物质。1
有机化学反应种类有机化学中类别较多,有自由基反应,离子型反应;亲电反应,亲核反应;硝化反应,卤化反应,磺化反应,氨化反应,酰化反应,氰化反应,加成反应,消去反应,取代反应,加聚反应,缩聚反应等。酸、碱
本词条内容贡献者为:
江长胜 - 教授、博导 - 西南大学
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国