

密度泛函理论 (英语:Density functional theory (DFT))是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一。
理论概述电子结构理论的经典方法,特别是Hartree-Fock方法和后Hartree-Fock方法,是基于复杂的多电子波函数的。密度泛函理论的主要目标就是用电子密度取代波函数做为研究的基本量。因为多电子波函数有3N个变量(N为电子数,每个电子包含三个空间变量),而电子密度仅是三个变量的函数,无论在概念上还是实际上都更方便处理。1
虽然密度泛函理论的概念起源于Thomas-Fermi模型,但直到Hohenberg-Kohn定理提出之后才有了坚实的理论依据。Hohenberg-Kohn第一定理指出体系的基态能量仅仅是电子密度的泛函。
Hohenberg-Kohn第二定理证明了以基态密度为变量,将体系能量最小化之后就得到了基态能量。
HK理论最初只适用于没有磁场存在的基态,现在已经被推广。最初的Hohenberg-Kohn定理仅仅指出了一一对应关系的存在,但是没有提供任何这种精确的对应关系。正是在这些精确的对应关系中存在着近似(这个理论可以被推广到时间相关领域,从而用来计算激发态的性质[6])。
密度泛函理论最普遍的应用是通过Kohn-Sham方法实现的。 在Kohn-Sham DFT的框架中,复杂的多体问题(由于处在一个外部静电势中的电子相互作用而产生的)被简化成一个没有相互作用的电子在有效势场中运动的问题。这个有效势场包括了外部势场以及电子间库仑相互作用的影响,例如交换和关联作用。处理交换关联作用是KS DFT的难点,目前尚没有精确求解交换相关能{\displaystyle E_{XC}}的方法。最简单的近似求解方法是局域密度近似(LDA)。LDA近似用均匀电子气来计算体系的交换能(均匀电子气的交换能是可以精确求解的),而采用对自由电子气进行拟合的方法来处理关联能。
自1970年以来,密度泛函理论在固体物理学计算中得到广泛的应用。多数情况下,与其它解决量子力学多体问题的方法相比,采用局域密度近似的密度泛函理论给出了非常令人满意的结果,同时固态计算相比实验的费用要少。尽管如此,人们普遍认为量子化学计算不能给出足够精确的结果,直到二十世纪九十年代,理论中所采用的近似被重新提炼成更好的交换关联作用模型。密度泛函理论是目前多种领域中电子结构计算的领先方法。 密度泛函理论尽管得到改进,但是描述分子间作用力,特别是范德华力,或者计算半导体的能隙还有一定困难。
早期模型: Thomas-Fermi 模型密度泛函理论可以上溯到由Thomas和Fermi在1920年代发展的Thomas-Fermi模型。他们将一个原子的动能表示成电子密度的泛函,并加上原子核-电子和电子-电子相互作用(两种作用都可以通过电子密度来表达)的经典表达来计算原子的能量。
Thomas-Fermi模型是很重要的第一步,但是由于没有考虑Hartree-Fock理论指出的原子交换能,Thomas-Fermi方程的精度受到限制。1928年保罗·狄拉克在该模型基础上增加了一个交换能泛函项。
然而,在大多数应用中Thomas-Fermi-Dirac理论表现得非常不够准确。其中最大的误差来自动能的表示,然后是交换能中的误差,以及对电子相关作用的完全忽略。
导出过程和表达式在通常的多体问题电子结构的计算中,原子核可以看作静止不动的(波恩-奥本海默近似),这样电子可看作在原子核产生的静电势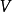 中运动。电子的定态可由满足多体薛定谔方程的波函数
中运动。电子的定态可由满足多体薛定谔方程的波函数 描述:
描述:
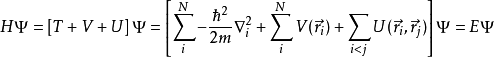 其中N为电子数目, U为电子间的相互作用势。算符 T和U 称为普适算符,它们在所有系统中都相同,而算符V则依赖于系统,为非普适的。可以看出,单粒子问题和比较复杂的多粒子问题的区别在于交换作用项U。目前有很多成熟的方法来解多体薛定谔方程,例如:物理学里使用的图形微扰理论和量子化学里使用的基于斯莱特行列式中波函数系统展开的组态相互作用(CI)方法。然而,这些方法的问题在于较大的计算量,很难用于大规模复杂系统的计算。
其中N为电子数目, U为电子间的相互作用势。算符 T和U 称为普适算符,它们在所有系统中都相同,而算符V则依赖于系统,为非普适的。可以看出,单粒子问题和比较复杂的多粒子问题的区别在于交换作用项U。目前有很多成熟的方法来解多体薛定谔方程,例如:物理学里使用的图形微扰理论和量子化学里使用的基于斯莱特行列式中波函数系统展开的组态相互作用(CI)方法。然而,这些方法的问题在于较大的计算量,很难用于大规模复杂系统的计算。
相比之下,密度函理论将含U的多体问题转化为不含U的单体问题上,成为解决此类问题的一个有效方法。在密度泛函理论中,最关键的变量为粒子密度 ,它由下式给出
,它由下式给出
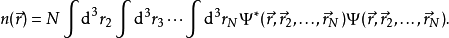 霍恩伯格和沃尔特·科恩在1964年提出,上面的关系可以反过来,即给出基态电子密度
霍恩伯格和沃尔特·科恩在1964年提出,上面的关系可以反过来,即给出基态电子密度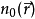 ,原则上可以计算出对应的基态波函数
,原则上可以计算出对应的基态波函数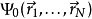 。也就是说,
。也就是说, 是
是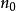 的唯一泛函,即
的唯一泛函,即
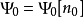 对应地,所有其它基态可观测量 O 均为 n0的泛函
对应地,所有其它基态可观测量 O 均为 n0的泛函
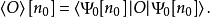 进而可以得出,基态能量也是n0的泛函
进而可以得出,基态能量也是n0的泛函
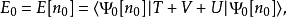 其中外势场的贡献
其中外势场的贡献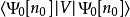 可以用密度表示成
可以用密度表示成
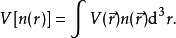
泛函T[n(r)]和 U[n]称为普适泛函,而V[n] 显然不是普适的,它取决于所考虑的系统。对于确定的系统,即V 已知,需要将泛函
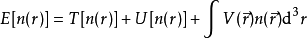
对于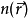 求极小值。这里假定能够得出 T[n(r)] 和U[n(r)]的表达式。对能量泛函求极值可以得到基态电子密度n0,进而求得所有基态可观测量。
求极小值。这里假定能够得出 T[n(r)] 和U[n(r)]的表达式。对能量泛函求极值可以得到基态电子密度n0,进而求得所有基态可观测量。
对能量泛函E[n(r)]求变分极值可以用不定算子的拉格朗日方法,这由科恩和沈吕九在1965年完成。这里我们使用如下结论:上面方程中的泛函可以写成一个无相互作用的体系的密度泛函
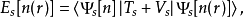 其中Ts为无相互作用的动能,Vs为粒子运动感受到的外势场。显然,
其中Ts为无相互作用的动能,Vs为粒子运动感受到的外势场。显然,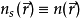 ,若Vs取为
,若Vs取为
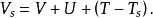 这样,可以解这个辅助的无相互作用体系的科恩-沈吕九方程
这样,可以解这个辅助的无相互作用体系的科恩-沈吕九方程
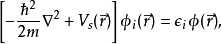 可以得到一系列的电子轨域,并由此求得原来的多体体系的电子密度
可以得到一系列的电子轨域,并由此求得原来的多体体系的电子密度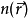
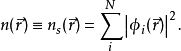 等效的单粒子势 Vs可以表示成
等效的单粒子势 Vs可以表示成
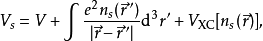
其中第二项为描述电子间库仑斥力的哈特里项,最后一项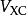 叫做交换关联势,包含所有多粒子的相互作用。由于哈特里项和交换关联项VXC都依赖于
叫做交换关联势,包含所有多粒子的相互作用。由于哈特里项和交换关联项VXC都依赖于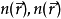 又依赖于
又依赖于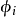 , 而
, 而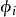 又依赖于Vs, 科恩-沈吕九方程的求解需要用自洽方法。通常首先假设一个初始的
又依赖于Vs, 科恩-沈吕九方程的求解需要用自洽方法。通常首先假设一个初始的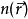 , 然后计算对应的Vs并求解科恩-沈吕九方程中的。进而可以计算出新的密度分布,并开始新一轮计算。此过程不断重复,直到计算结果收敛。
, 然后计算对应的Vs并求解科恩-沈吕九方程中的。进而可以计算出新的密度分布,并开始新一轮计算。此过程不断重复,直到计算结果收敛。
本词条内容贡献者为:
杜强 - 高级工程师 - 中国科学院工程热物理研究所
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国