

基本原理
原子吸收光谱法 (AAS)是利用气态原子可以吸收一定波长的光辐射,使原子中外层的电子从基态跃迁到激发态的现象而建立的。由于各种原子中电子的能级不同,将有选择性地共振吸收一定波长的辐射光,这个共振吸收波长恰好等于该原子受激发后发射光谱的波长。当光源发射的某一特征波长的光通过原子蒸气时,即入射辐射的频率等于原子中的电子由基态跃迁到较高能态(一般情况下都是第一激发态)所需要的能量频率时,原子中的外层电子将选择性地吸收其同种元素所发射的特征谱线,使入射光减弱。特征谱线因吸收而减弱的程度称吸光度A,在线性范围内与被测元素的含量成正比: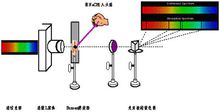
A=KC
式中K为常数;C为试样浓度;K包含了所有的常数。此式就是原子吸收光谱法进行定量分析的理论基础
由于原子能级是量子化的,因此,在所有的情况下,原子对辐射的吸收都是有选择性的。由于各元素的原子结构和外层电子的排布不同,元素从基态跃迁至第一激发态时吸收的能量不同,因而各元素的共振吸收线具有不同的特征。由此可作为元素定性的依据,而吸收辐射的强度可作为定量的依据。AAS现已成为无机元素定量分析应用最广泛的一种分析方法。该法主要适用样品中微量及痕量组分分析。
谱线轮廓原子吸收光谱线并不是严格几何意义上的线,而是占据着有限的相当窄的频率或波长范围,即有一定的宽度。原子吸收光谱的轮廓以原子吸收谱线的中心波长和半宽度来表征。中心波长由原子能级决定。半宽度是指在中心波长的地方,极大吸收系数一半处,吸收光谱线轮廓上两点之间的频率差或波长差。半宽度受到很多实验因素的影响。影响原子吸收谱线轮廓的两个主要因素: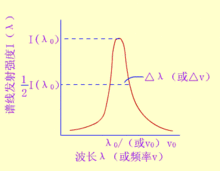
1、多普勒变宽。多普勒宽度是由于原子热运动引起的。从物理学中已知,从一个运动着的原子发出的光,如果运动方向离开观测者,则在观测者看来,其频率较静止原子所发的光的频率低;反之,如原子向着观测者运动,则其频率较静止原子发出的光的频率为高,这就是多普勒效应。原子吸收分析中,对于火焰和石墨炉原子吸收池,气态原子处于无序热运动中,相对于检测器而言,各发光原子有着不同的运动分量,即使每个原子发出的光是频率相同的单色光,但检测器所接受的光则是频率略有不同的光,于是引起谱线的变宽。
2、碰撞变宽。当原子吸收区的原子浓度足够高时,碰撞变宽是不可忽略的。因为基态原子是稳定的,其寿命可视为无限长,因此对原子吸收测定所常用的共振吸收线而言,谱线宽度仅与激发态原子的平均寿命有关,平均寿命越长,则谱线宽度越窄。原子之间相互碰撞导致激发态原子平均寿命缩短,引起谱线变宽。碰撞变宽分为两种,即赫鲁兹马克变宽和洛伦茨变宽。
赫鲁兹马克变宽是指被测元素激发态原子与基态原子相互碰撞引起的变宽,称为共振变宽,又称赫鲁兹马克变宽或压力变宽。在通常的原子吸收测定条件下,被测元素的原子蒸气压力很少超过10-3mmHg,共振变宽效应可以不予考虑,而当蒸气压力达到0.1mmHg时,共振变宽效应则明显地表现出来。洛伦茨变宽是指被测元素原子与其它元素的原子相互碰撞引起的变宽,称为洛伦茨变宽。洛伦茨变宽随原子区内原子蒸气压力增大和温度升高而增大。
除上述因素外,影响谱线变宽的还有其它一些因素,例如场致变宽、自吸效应等。但在通常的原子吸收分析实验条件下,吸收线的轮廓主要受多普勒和洛伦茨变宽的影响。在2000-3000K的温度范围内,原子吸收线的宽度约为10-3-10-2nm。
仪器结构原子吸收光谱仪由光源、原子化系统、分光系统、检测系统等几部分组成。通常有単光束型和双光束型两类。这种仪器光路系统结构简单,有较高的灵敏度,价格较低,便于推广,能满足日常分析工作的要求,但其最大的缺点是,不能消除光源被动所引起的基线漂移,对测定的精密度和准确度有意境的影响。1
1、 光源。光源的功能是发射被测元素的特征共振辐射。对光源的基本要求是:发射的共振辐射的半宽度要明显小于吸收线的半宽度;辐射强度大、背景低,低于特征共振辐射强度的1%;稳定性好,30分钟之内漂移不超过1%;噪声小于0.1%;使用寿命长于5安培小时。空心阴极放电灯是能满足上述各项要求的理想的锐线光源,应用最广。
2、原子化器。其功能是提供能量,使试样干燥,蒸发和原子化。 在原子吸收光谱分析中,试样中被测元素的原子化是整个分析过程的关键环节。原子化器主要有四种类型火焰原子化器、石墨炉原子化器、氢化物发生原子化器及冷蒸气发生原子化器。实现原子化的方法,最常用的有两种:
火焰原子化法:是原子光谱分析中最早使用的原子化方法,至今仍在广泛地被应用;
非火焰原子化法,其中应用最广的是石墨炉电热原子化法。
3、分光器。它由入射和出射狭缝、反射镜和色散元件组成,其作用是将所需要的共振吸收线分离出来。分光器的关键部件是色散元件,商品仪器都是使用光栅。原子吸收光谱仪对分光器的分辨率要求不高,曾以能分辨开镍三线Ni230.003、Ni231.603、Ni231.096nm为标准,后采用Mn279.5和279.8nm代替Ni三线来检定分辨率。光栅放置在原子化器之后,以阻止来自原子化器内的所有不需要的辐射进入检测器。
4、检测系统。原子吸收光谱仪中广泛使用的检测器是光电倍增管,一些仪器也采用CCD作为检测器。
干扰效应原子吸收光谱分析法与原子发射光谱分析法相比,尽管干扰较少并易于克服,但在实际工作中干扰效应仍然经常发生,而且有时表现得很严重,因此了解干扰效应的类型、本质及其抑制方法很重要。原子吸收光谱中的干扰效应一般可分为四类:物理干扰、化学干扰、电离干扰和光谱干扰。
物理干扰及其抑制物理干扰指试样在前处理、转移、蒸发和原子化的过程中,试样的物理性质、温度等变化而导致的吸光度的变化。物理干扰是非选择性的,对溶液中各元素的影响基本相似。
削除和抑制物理干扰常采用如下方法:
(1) 配制与待测试样溶液相似组成的标准溶液,并在相同条件下进行测定。如果试样组成不详,采用标准加入法可以削除物理干扰。
(2) 尽可能避免使用粘度大的硫酸、磷酸来处理试样;当试液浓度较高时,适当稀释试液也可以抑制物理干扰。
化学干扰及其抑制化学干扰是指待测元素在分析过程中与干扰元素发生化学反应,生成了更稳定的化合物,从而降低了待测元素化合物的解离及原子化效果,使测定结果偏低。这种干扰具有选择性,它对试样中各种元素的影响各不相同。化学干扰的机理很复杂,
消除或抑制其化学干扰应该根据具体情况采取以下具体措置措施:
1、加入干扰抑制剂
(1) 加入稀释剂 加入释放剂与干扰元素生成更稳定或更难挥发的化合物,从而使被测定元素从含有干扰元素的化合物中释放出来。
(2) 加入保护剂 保护剂多数是有机络合物。它与被测定元素或干扰元素形成稳定的络合物,避免待测定元素与干扰元素生成难挥发化合物。
(3) 加入缓冲剂 有的干扰,当干扰物质达到一定浓度时,干扰趋于稳定,这样,把被测溶液和标准溶液加入同样量的干扰物质时,干扰物质对测定就不会发生影响。
2、选择合适的原子化条件
提高原子化温度,化学干扰一般会减小,使用高温火焰或提高石墨炉原子化温度,可使难解离的化合物分解。
3、加入基体改进剂
用石墨炉原子化时,在试样中加入基体改进剂,使其在干燥或灰化阶段与试样发生化学变化,其结果可能增强基体的挥发性或改变被测元素的挥发性,使待测元素的信号区别于背景信号。
当以上方法都未能消除化学干扰时,可采用化学分离的方法,如溶剂萃取、离子交换、沉淀分离等方法。
电离干扰及其抑制电离干扰是指待测元素在高温原子化过程中,由于电离作用而使参与原子吸收的基态原子数目减少而产生的干扰。
为了抑制这种电离干扰,可加入过量的消电离剂。由于消电离剂在高温原子化过程中电离作用强于待测元素,它们可产生大量自由电子,使待测元素的电离受到抑制,从而降低或消除了电离干扰。
光谱干扰及其抑制光谱干扰是指在单色器的光谱通带内,除了待测元素的分析线之外,还存在与其相邻的其他谱线而引起的干扰,常见的有以下三种。
1、吸收线重叠
一些元素谱线与其他元素谱线重叠,相互干扰。可另选灵敏度较高而干涉少的分析线抑制干扰或采用化学分离方法除去干扰元素。
2、光谱通带内的非吸收线
这是与光源有关的光谱干扰,即光源不仅发射被测元素的共振线,往往发射与其邻近的非吸收线。对于这些多重发射,被测元素的原子若不吸收,它们被监测器检测,产生一个不变的背景型号,使被测元素的测定敏感度降低;若被测元素的原子对这些发射线产生吸收,将使测定结果不正确,产生较大的正误差。
消除方法,可以减小狭缝宽度,使光谱通带小到可以阻挡多重发射的谱线,若波长差很小,则应另选分析线,降低灯电流也可以减少多重发射。
3、背景干扰和抑制
背景干扰包括分子吸收、光散射等。
分子吸收是原子化过程中生成的碱金属和碱土金属的卤化物、氧化物、氢氧化物等的吸收和火焰气体的吸收,是一种带状光谱,会在一定波长范围内产生干扰。
光散射是原子化过程中产生的微笑固体颗粒使光产生散射,吸光度增加,造成假吸收。波长越短,散射影响越大。
背景干扰都使吸光度增大,产生误差。石墨炉原子化法背景吸收干扰比火焰原子化法来得严重,有时不扣除背景会给测定结果带来较大误差。
目前用于商品仪器的背景矫正方法主要是氘灯扣除背景、塞曼效应扣除背景。1
主要特点优越性原子吸收光谱法该法具有检出限低(火焰法可达μg/cm–3级)准确度高(火焰法相对误差小于1%),选择性好(即干扰少)分析速度快,应用范围广(火焰法可分析30多种/70多种元素,石墨炉法可分析70多种元素,氢化物发生法可分析11种元素)等优点2。
1 选择性强。这是因为原子吸收带宽很窄的缘故。因此,测定比较快速简便,并有条件实现自动化操作。在发射光谱分析中,当共存元素的辐射线或分子辐射线不能和待测元素的辐射线相分离时,会引起表观强度的变化。
而对原子吸收光谱分析来说:谱线干扰的几率小,由于谱线仅发生在主线系,而且谱线很窄,线重叠几率较发射光谱要小得多,所以光谱干扰较小。即便是和邻近线分离得不完全,由于空心阴极灯不发射那种波长的辐射线,所以辐射线干扰少,容易克服。在大多数情况下,共存元素不对原子吸收光谱分析产生干扰。在石墨炉原子吸收法中,有时甚至可以用纯标准溶液制作的校正曲线来分析不同试样。
2、灵敏度高。原子吸收光谱分析法是目前最灵敏的方法之一。火焰原子吸收法的灵敏度是ppm到ppb级,石墨炉原子吸收法绝对灵敏度可达到10-10~10-14g。常规分析中大多数元素均能达到ppm数量级。如果采用特殊手段,例如预富集,还可进行ppb数量级浓度范围测定。由于该方法的灵敏度高,使分析手续简化可直接测定,缩短分析周期加快测量进程;由于灵敏度高,需要进样量少。无火焰原子吸收分析的试样用量仅需试液5~100?l。固体直接进样石墨炉原子吸收法仅需0.05~30mg,这对于试样来源困难的分析是极为有利的。譬如,测定小儿血清中的铅,取样只需10?l即可。
3 分析范围广。发射光谱分析和元素的激发能有关,故对发射谱线处在短波区域的元素难以进行测定。另外,火焰发射光度分析仅能对元素的一部分加以测定。例如,钠只有1%左右的原子被激发,其余的原子则以非激发态存在。
在原子吸收光谱分析中,只要使化合物离解成原子就行了,不必激发,所以测定的是大部分原子。应用原子吸收光谱法可测定的元素达73种。就含量而言,既可测定低含量和主量元素,又可测定微量、痕量甚至超痕量元素;就元素的性质而言,既可测定金属元素、类金属元素,又可间接测定某些非金属元素,也可间接测定有机物;就样品的状态而言,既可测定液态样品,也可测定气态样品,甚至可以直接测定某些固态样品,这是其他分析技术所不能及的。
4、抗干扰能力强。第三组分的存在,等离子体温度的变动,对原子发射谱线强度影响比较严重。而原子吸收谱线的强度受温度影响相对说来要小得多。和发射光谱法不同,不是测定相对于背景的信号强度,所以背景影响小。在原子吸收光谱分析中,待测元素只需从它的化合物中离解出来,而不必激发,故化学干扰也比发射光谱法少得多。
5、精密度高。火焰原子吸收法的精密度较好。在日常的一般低含量测定中,精密度为1~3%。如果仪器性能好,采用高精度测量方法,精密度为
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国