

内容一览:了解全局晶体对称性并分析等变信息,对于预测材料性能至关重要,但现有的、基于卷积网络的算法尚且无法完全实现这些需求。针对于此,中山大学的李华山、王彪课题组,开发了一款名为 SEN 的机器学习模型,准确感知了固有晶体对称性和材料结构团簇之间的相互作用。
关键词:材料性能预测 深度学习 MP 数据库
作者 | 李宝珠
编辑 | 三羊
晶体对称性对于研究材料的物理性质、理解晶体结构、设计新材料以及进行 X 射线衍射等实验具有关键作用。了解晶体对称性有助于简化分析,更好地理解材料属性,并提高材料性能的计算效率。更重要的是,晶体对称性还可以直接影响材料的电荷分布、光学性质、磁性质等物理特性。
近年来,基于统计机制的机器学习已经得到了广泛应用,而从机器学习的角度来看,晶体对称性可以看作是材料的不变性与等变形,但目前现有的、基于高级图网络的晶体材料机器学习算法很难编码复杂的材料不变性和等变性。
此外,堆叠式胶囊自编码器 (Stacked Capsule Autoencoder,SCAE) 虽然也可以直接从原始数据中提取空间对称性特征,但传统的胶囊模型仍无法分析复杂材料系统结构-性能之间的关系。
针对以上挑战,中山大学李华山、王彪课题组开发了一套名为 SEN(symmetry-enhanced equivariance network,对称增强等变网络 )的机器学习模型,克服了基于卷积的算法在高对称空间群中性能不佳的问题,并在所有空间群中实现了高精度的材料性能预测。目前,相关成果已经发表于「Nature Communications」。
相关成果已经发表于「Nature Communications」
获取论文:
https://www.nature.com/articles/s41467-023-40756-2
01 数据集:MP

数据库中的 6,027 个晶体材料
研究人员基于化学环境的概念和图模型的表示方法提取了晶体材料的特征,以目标原子截断半径内的周围原子和键来定义其化学环境,并从用于材料分析的开源 Python 数据库——Materials Project 中提取了每个原子周围的原子类型、原子连接性和键长。
据悉,本研究中用于预测带隙和形成能的数据集来自 Materials Project 数据库,带隙和形成能的数据集分别包含 6,027 (按 8:1:1 的比例分为训练集、验证集和测试集)和 30,000 种材料。这两个数据集由 64 个元素组成,涵盖了周期表内除惰性气体组、镧系元素、锕系元素和放射性元素外的元素。
研究人员通过密度泛函理论(DFT)计算,对 Materials Project 数据库中的 6,027 个晶体材料组成的数据集进行了预测,并基于预测结论对 SEN 模型的性能进行了检验。
本研究中使用的晶体对称性和化学环境数据可从 Zenodo 数据库中获取。
访问链接:
https://doi.org/10.5281/zenodo.8142678
02 模型架构:3 个模块统一训练
如下图所示,SEN 模型采用了复杂的深度学习架构,包含特征提取 (FE)、对称性感知 (SP) 和属性预测 (PP) 模块。

SEN 架构由特征提取、对称感知和属性预测模块组成
在本次研究中,研究团队通过对 3 个模块的统一训练,实现了对多种材料特性的准确预测,并通过 SEN 模型描述了原子之间的相互作用。
首先,特征提取模块感知输入的原子和化学键数据,输入数据包括了靶材料原始单元中 N 原子和 M 键的信息。最后,通过高通量筛选过程,构建了包括化学计量、晶体结构、原子信息和键信息的材料数据集。
以材料数据集作为 SEN 模型的唯一输入数据,研究人员基于结构数据和化学计量数据,同时计算出了原子化学环境向量 VmA,以及元素权重向量 VmE。
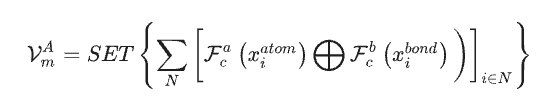
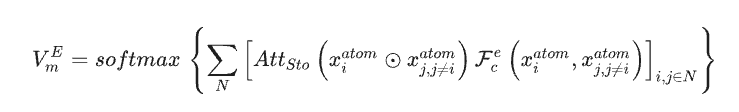
经多层感知器激活后,元素权重向量被转换为相应原子的概率向量。研究人员进而通过原子化学环境向量和元素权重向量之间的 element-wise operation,更新了所有原子级别的相关性,从而能通过 LSTM-attention 层获得了材料的化学环境矩阵。
其次,该研究创新性地将胶囊机制 (capsule mechanism) 应用于材料属性预测,通过基于胶囊机制设计的对称性感知模块,将材料化学环境转换为由对称算子、卷积材料化学环境和存在值组成的材料胶囊,以感知并保留晶体对称性。进而,通过在材料的化学环境矩阵上进行对称运算,可以将不同的对称图案推广到晶体胶囊中。
最后,在属性预测方面,SEN 模型通过基于 MLP 的映射函数预测目标材料性质。
03 SEN 模型高精度预测材料属性
结论一:SEN 模型准确感知原子相互作用信息
为了验证特征提取模块的有效性,研究人员训练了 SEN 预测晶体材料带隙的能力,直到平均绝对误差 (MAE) 低于 0.15 eV,然后分析了特征提取模块产生的化学环境中间数据。
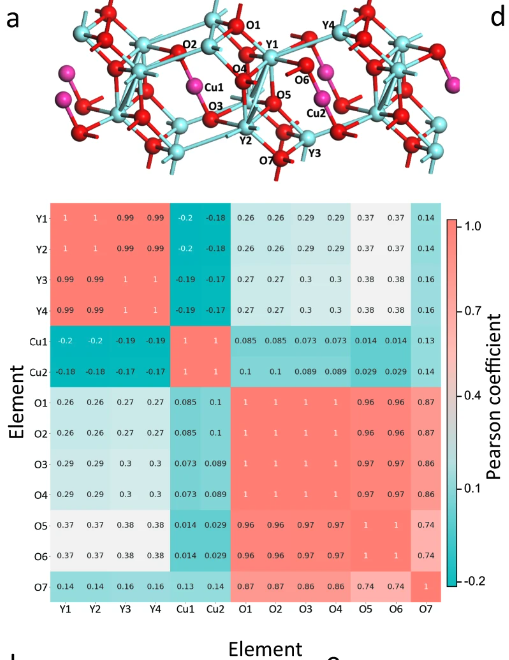
基于原子的化学环境相关性分析
具体而言,研究人员提取了 Y4Cu2O7 的原胞中每个原子的化学环境矩阵。计算了原子矩阵之间的 Pearson 系数,生成了上图所示的相关性分析图。与不同元素组的原子相比,同一元素组内的原子之间的 Pearson 系数要大得多,因此可以清楚地区分出 Y4Cu2O7 中的 3 个元素组。
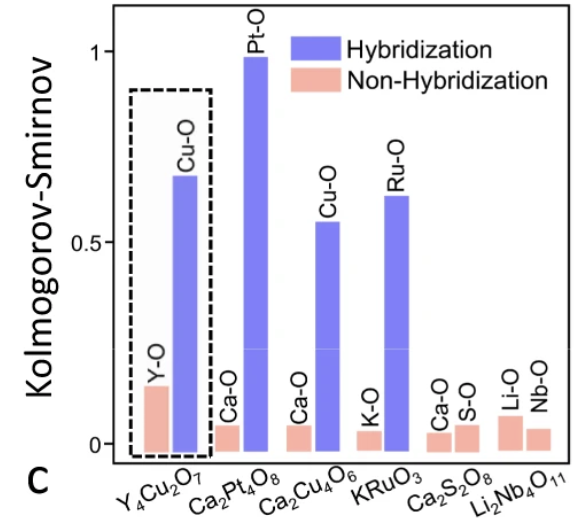
通过 SEN 模型学习了六种材料的原子相关性

如上图所示,SEN 模型已经学习并编码了原子相互作用信息,并成功地检测到了杂化现象,这对于电子性质的预测具有重要意义。
结论二:SEN 模型预测性能优于 MegNet
为了研究在 SEN 模型中从化学环境到材料性质的映射,研究人员从 MP 数据库中选择了五种材料——Be(6)Ni(2)、 Sr(4)Ge(2)S(8)、 Li(2)V(2)F(12)、 CsAsF(6)、 BaB(2)F(8),其带隙分别为 0 eV、 3.25 eV、 4.86 eV、 7.24 eV 及 10.12 eV。
观察得知,带隙与材料化学环境的 PDF(概率密度函数)之间存在强相关性,即随着带隙的增大,PDF 逐渐扩散。整个数据集从材料化学环境到带隙的投影如下图所示,6,027 个晶体材料均匀分布在主特征空间,而带隙的变化在整个空间上是连续、单调的。
6027 种材料的 2D t-SNE 图,圆的颜色表示带隙值
为了验证机器学习模型所学习的特征-属性关系符合基本物理原理,研究人员生成了 Ca-O-X 材料的化学环境 2D t-SNE 图,并调查了各种材料特征(成分、点群、自旋极化等),最终发现,材料带隙取决于复杂的材料特征,不能简单地由任一关键因素来预测。
尽管如此,SEN 模型在带隙预测方面还是取得了显著的提升。在对测试数据集中的材料带隙进行预测时,SEN 模型的均方误差 (MAE) 为 0.25 eV,与具有 MLP、DenseNet、TFN、SE(3) 和 EGNN 模块的模型在测试数据集上获得的 MAE 相比,有显著改进。
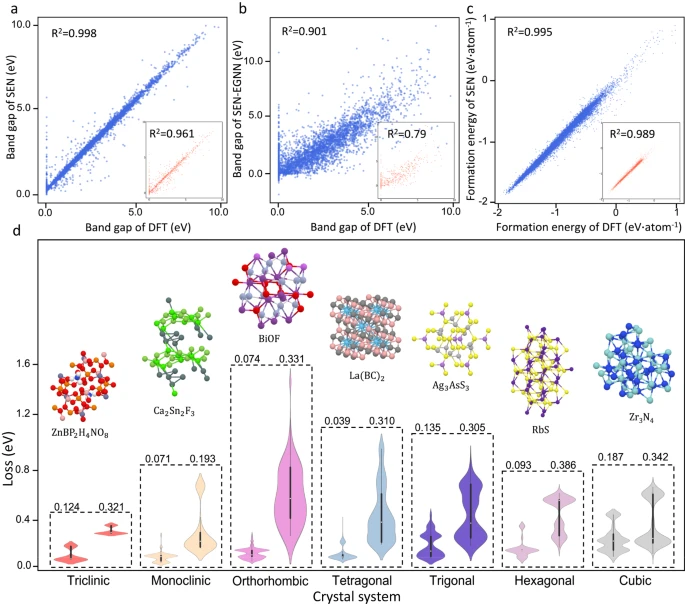
不同对称度晶体材料性能的预测
如上图 d 所示,研究人员对比检验了 SEN 模型和 MegNet21 模型(通用材料网络模型)对不同晶体系统的预测质量,进一步揭示了对称感知对材料性能预测的显著影响。从误差分布图来看,SEN 模型的预测性能在所有晶体系统中均优于 MegNet。
此外,SEN 模型通过感知全晶体对称性,大幅降低了有效特征维数。这一特征清除过程减轻了过拟合问题,并加强了从材料特征到属性的映射。
论文显示,SEN 模型预测带隙和形成能的平均绝对误差分别比常见机器学习模型低约 22.9% 和 38.3%。
04 AI 推动材料产业变革发展
长久以来,新材料的设计、研发以及材料性能的改革是牵引科技进步的拉力之一,在电子、能源、医疗、航空航天等诸多领域发挥着重要作用。但传统的材料研发过程往往需要大量实验来不断修正性能,提高可行性,这一过程漫长且需要耗费极大的人力、财力。
而随着 AI 的加速应用,AI for Science 得到了越来越多的关注,其与材料的结合也成为了越来越多学者、企业的探索新方向。一方面,AI 可以分析大量数据,并进行模拟预测,从而加速新材料的发现与性能优化;另一方面,材料学科也成为了机器学习、自然语言处理、高性能计算等 AI 关键技术的重要落脚点。
可以说,AI 正在润物细无声地改变着新材料的设计与应用。未来,随着更强大的 AI 模型持续迭代,加之数据共享之下材料数据库的更新扩充,AI 势必将进一步推动新材料的诞生。



 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国