

导读
近日,Science上同一天上线了两篇C-H活化领域文章。
一篇是碳氢键活化领域大咖、Scripps研究所的余金权教授课题组新发表的Science,报道了在手性双功能噁唑啉-吡啶酮配体下,实现了游离环烷烃羧酸的对映选择性钯催化远程γ-C-H(杂)芳基化。该反应可同时构建γ-叔碳和α-季碳手性中心,ee高达>99%,获得了多种环状手性合成子和生物活性分子。通过使用具有相反构型的手性配体还可以构建含有三个手性中心的碳环,实现两个亚甲基C-H键的顺序对映选择性编辑。还可实现对映选择性远程 δ-C-H(杂)芳基化,以构建δ-手性中心。文章链接DOI:10.1126/science.ado1246
另一篇是奥地利维也纳大学的Nuno Maulide教授课题组报道了一种通过远程质子消除形成碳-碳σ键的方法,这是一种通过五个碳-碳键的远端酸化实现的独特的C-H键活化模式,远程质子消除法在环癸基阳离子中的应用为合成十氢萘化合物提供了一种有吸引力的方法。这种转变是区域收敛(regioconvergent)的,不需要导向基或贵金属,并表现出精准的位点选择性。作者通过深入的计算研究阐明了反应机理,此外,还描述了通过氢原子转移介导的差向异构化,十氢萘产物表现出完全的立体异构体富集。文章链接DOI:10.1126/science.adi8997

1、余金权教授课题组Science:环烷烃羧酸的对映选择性远端亚甲基C-H(杂)芳基化
图片来源:Science
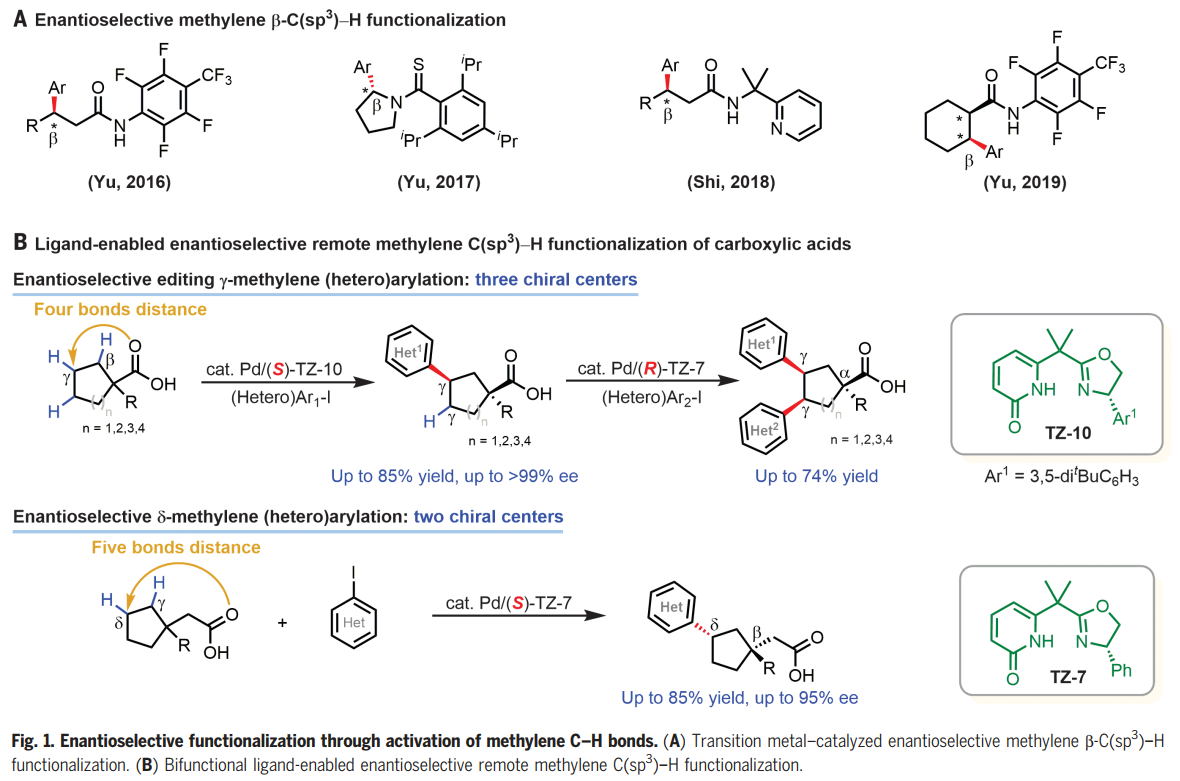
过渡金属催化的对映选择性C-H活化反应已成为一种强大且通用的方法,用于不对称合成点、轴和平面手性的分子。在手性双功能单N保护氨基酸(MPAA)配体及相关手性配体下,通过选择性活化其中一个甲基以及亚甲基的直接不对称活化,可实现偕二甲基的去对称化。然而,除了环丙基和环丁基底物之外,对映选择性亚甲基C-H活化方法仍然仅限于含有更强的外源导向基的底物。此外,这些方法仅限于近端手性中心距导向原子最多三个键的底物(Fig. 1A)。考虑到大多数生物活性天然产物和药物分子都含有碳环,这有利于限制构象灵活性,从而改善ADME特性,如口服生物利用度,而多功能环烷烃羧酸起始原料的γ-和δ-C-H官能团化对于合成富含sp3碳的生物活性分子特别有价值。在这里,作者发展了手性双功能噁唑啉-吡啶酮配体,其能够实现多种无环羧酸的对映选择性远程γ-C-H(杂)芳基化,构建γ-叔碳手性中心,同时α-季碳中心去对称化高达>99% ee,为各种环状手性合成子和生物活性分子提供了高度对映选择性途径。通过使用具有相反构型的手性配体在这些空间拥挤的碳环中构建第三个手性中心,证明了两个亚甲基C-H键的顺序对映选择性编辑。手性Pd/噁唑啉-吡啶酮催化剂还能够进行对映选择性δ-C-H(杂)芳基化(Fig. 1B)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
选择性地形成羰基α-、β-、γ-和δ-位手性中心的反应是不对称有机合成中最理想和最通用的转化之一。化学家们已经报道了许多能够高选择性构建α-和β-手性中心的对映选择性反应。相比之下,催化对映选择性形成羰基的γ-和δ-手性中心仍然极具挑战。受作者之前通过C-H活化构建α-和β-手性中心以及开发碳环外消旋γ-C-H 芳基化的成功的鼓舞(Science, 2018, DOI: 10.1126/science.aao4798;Nature, 2023, 618, 519-525),余金权团队寻求开发能够对映选择性远程催化γ-和δ-C-H功能化的手性Pd催化剂,有望解决这一未满足的需求。
图片来源:Science
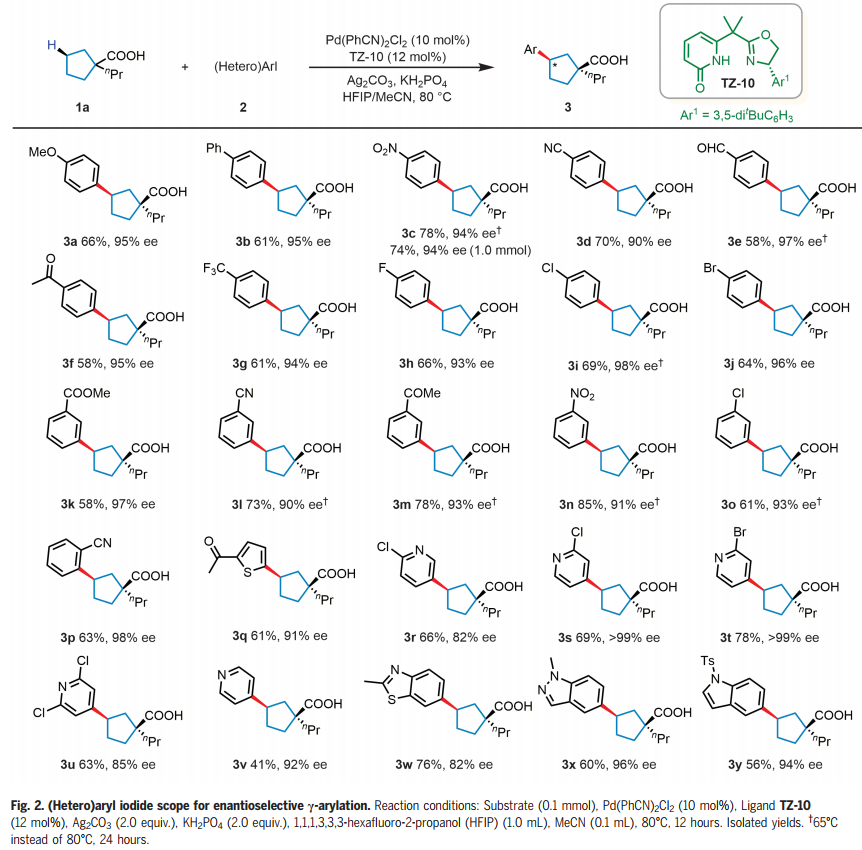
鉴于环系统在有机化学中的重要性,选择1-丙基环戊烷-1-羧酸(1a)和4-碘苯甲酸甲酯(2z)作为反应开发和配体设计的模型底物。手性吡啶酮-磺酰胺配体(L7)和联萘酚衍生的吡啶-吡啶酮配体(L8)以中等产率(45%-57%)提供所需的芳基化产物,表明吡啶酮单元对于激活亚甲基 C-H键至关重要。作者合成了一系列手性双功能噁唑啉-吡啶酮配体,它们可以作为五元(TZ-1至4)或六元(TZ-5至12)螯合物与Pd(II)配位,事实证明,六元螯合物(TZ-5 至 12)则提供中等至良好的产率。噁唑啉环上芳基取代基的存对于在该反应中获得立体控制至关重要,芳基化类似物(TZ-7至10)提供具有优异水平对映选择性(85-97%ee)的γ-芳基化产物。经过对反应的广泛优化,六元螯合噁唑啉-吡啶酮配体 TZ-10被确定为最佳配体,形成γ-芳基化产物,收率74%,ee 97%。
有了最佳的配体和条件,作者开始研究环戊基羧酸(1a)反应中芳基和杂芳基碘化物的范围(Fig. 2)。芳基碘化物上的各种对位取代基,从给电子(OMe)到吸电子(包括NO2、CN和CF3)都是相容的,得到相应的γ-芳基化酸(3a-g),产率58%-78%,具有优异的对映选择性(90%-97% ee),仅检测到顺式产物。还可以耐受卤素取代基,如氟、氯、溴,以64-69%的产率和93-98% ee得到所需产物(3h-j)。带有间位或邻位取代基的芳基碘化物也表现良好,以中等至良好的产率和90%-98% ee形成所需产物。接下来考察了杂芳基化反应,多种2-取代杂芳基碘化物能够以中等收率和优异的ee形成相应的产物。即使是4-碘吡啶,也能以41%的产率和92% ee提供相应的产物(3v)。
图片来源:Science
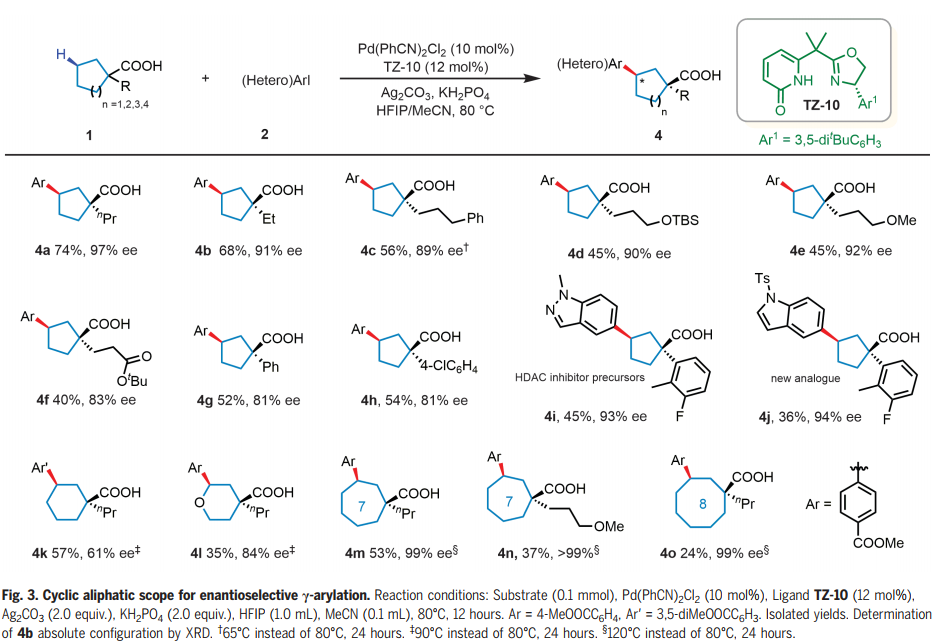
接下来,作者研究了环烷羧酸的范围(Fig. 3)。在羧酸的α-位带有醚或酯官能团的各种烷基取代物是相容的,以40-74%的产率和83-97% ee 提供偶联产物(4a-f)。羧酸α-位的芳基也可以耐受,提供36-54%产率和81-94% ee 的芳基化产物,质量平衡高于85%,其中环烷羧酸回收率为30-50%。市售的α-芳基环戊烷甲酸成功与杂芳基碘化物偶联,生成了HDAC抑制剂前体(4i)及其新的类似物(4j),收率36-45%,ee 93-94%。与以前繁琐的方法(十步,产率和手性分辨率较低)相比,本文的反应为合成这些药物分子提供了一条显著改进的途径。六元环烷烃和杂环羧酸提供了具有中等到高对映选择性的γ-芳基化产物(4k-1)。七(4m-n)和八(4o)元环也顺利反应,提供了目标γ-芳基化产物,其ee率为99%。
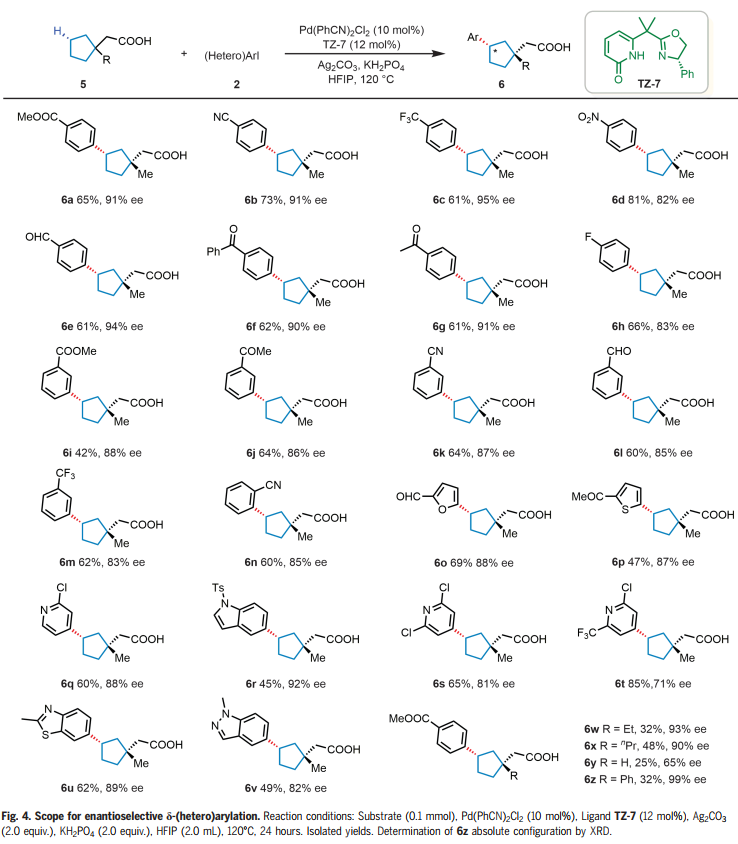
受到γ-亚甲基C-H芳基化结果的鼓舞,作者想知道这些手性双功能噁唑啉-吡啶酮配体是否也可以实现更远端δ-亚甲基C-H键的对映选择性芳基化。选择2-(1-甲基环戊基)乙酸(5a)与4-碘苯甲酸甲酯(2z)作为模型底物。通过对反应条件的广泛优化,将产率提高到64%,同时保持91% ee,仅检测到顺式产物。接下来用底物5a来考察芳基和杂芳基碘化物的范围(Fig. 4)。对位、间位和邻位的各种不同官能团是兼容的,以中等至良好的产率(42%-81%)和82%-95% ee提供δ-芳基化产物(6a-n)。各种含有呋喃基、噻吩基、苯并噻唑基、吲哚基和吡啶基的杂芳基碘化物成功偶联,得到所需产物(6o-v),收率45%-85%,ee 71%-92%。带有β-乙基和β-丙基取代基的羧酸也表现良好,产物 (6w-x) 32%-48%的产率和90%-93% ee。这一单一操作同时在碳环上构建 β-季碳和δ-手性中心,而使用以前的方法很难完成这一任务。
图片来源:Science
最近,构建1,2-二芳基取代的碳环的重要性在许多药物发现项目中变得越来越明显。作者设想了一种连续的对映选择性C-H芳基化过程,该过程可以为制备多种1,2-芳基化碳环提供一种通用的方法(Fig. 5A)。通过具有相反构型的手性配体(R)-TZ-7,可以以30%-74%的产率获得二次芳基化和杂芳基化产物(7a-d)。仅观察到单一顺式二芳基化非对映异构体。更具挑战性的二杂芳基取代的碳环(7e)也以32%的产率制备。衍生自孕烯醇酮和半乳糖的芳基碘化物也是相容的,以64%-67%的产率提供所需的偶联产物(8a-b),dr > 20:1(Fig. 5B)。

图片来源:Science
余金权团队用手性双功能噁唑啉-吡啶酮配体(TZ-7和TZ-10)实现了环戊基、环己基、环庚基和环辛基羧酸的对映选择性远程γ-亚甲基 C-H(杂)芳基化。该方法将扩展到其他对映选择性C-H 转化,包括烷基化和乙烯基化。
2、Nuno Maulide课题组Science:远程质子消除,通过远端酸化实现 C-H 激活

有机分子C-H键功能化方法的发展已经改变了化学合成领域。C-H活化有助于将合成计划与分子中存在的官能团的固有反应性分离,从而实现更有效的合成途径,以得到有价值的目标,如药物和农用化学品等。
在过去的50年里,一系列的研究已经发现了许多能够激活C-H键的化学系统,但是能够激活C-H键裂解的基元步骤类型相对较少。当从机理的角度看待文献主体时,只有四类C-H键激活涵盖了大多数例子。这些包括(i)金属的氧化加成,(ii)使用弱碱的定向金属化,(iii) 氢原子提取,以及(iv)卡宾或者氮宾插入(Fig. 1A)。通过以前未探索的机制操作的C-H激活过程的识别可以被视为该领域持续发展的关键。 化学加——科学家创业合伙人,欢迎下载化学加APP关注。
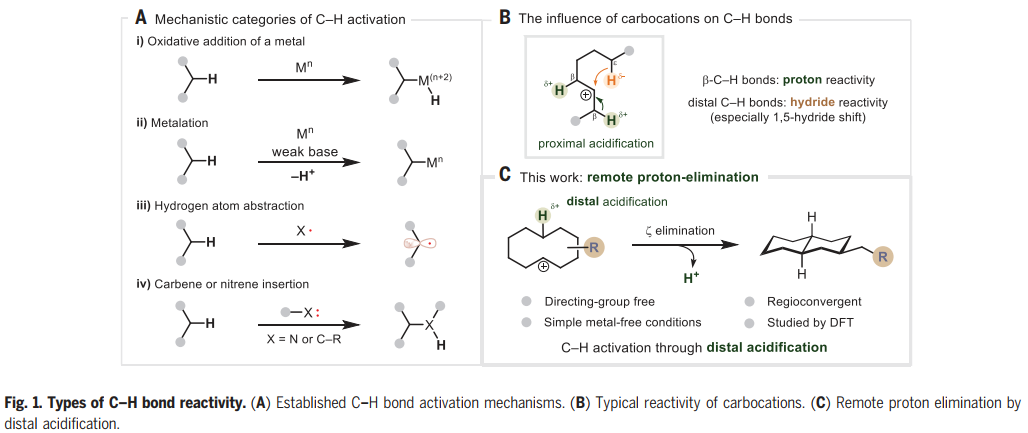
图片来源:Science
为了阐明C-H活化的不同模式,Nuno Maulide教授课题组报道了碳阳离子的远程消除反应,导致C-C键的形成(Fig. 1C)。众所周知的碳阳离子倾向于酸化近端β-C-H键,同时诱导远端C-H键的氢化物反应性,而这一过程涉及相反的极化情况,即酸化远端C-H键。环癸基碳阳离子的消去可以从预期的E1途径(通过β-消去产生新的π-C=C键)转移到涉及在五个键分隔的碳之间形成新的σ-C-C键的途径(即ζ消去)。远程消去被证明是合成十氢萘的有效策略,依赖于桥接C-C键的非典型逆合成断开,而不是一个或多个非桥接键(如Robinson环化)。
文章的灵感来自于1955年Cope小组的一份报告,该报告描述了用2-萘磺酸处理反式环癸烯形成少量十氢萘。反应的主要产物是顺-环癸烯,可能是由E1 β消除产生的。由于中环张力的减轻,十氢萘的形成在热力学上应该是有利的,Cope的结果表明,动力学控制有利于β-消除。Nuno Maulide教授课题组推测β-消除在酸性条件下是可逆的,而远程(ζ)消除是不可逆的。鉴于β-消除的可逆性,作者认为,可以确定一组热力学条件来有选择地促进ζ消除(Fig. 2A)。
这一评估被证明是正确的,并且能够开发一套热力学条件来选择性地促进远程消除,在65 °C下使用三氟甲磺酸(TfOH)与六氟异丙醇(HFIP)的组合(Fig. 2B)。传统的无氟溶剂不如HFIP,这一效应在其他几项涉及Brønsted酸介导反应的研究中也观察到了。在较低的温度下(0 ℃为1.1),得到环癸烯的混合物,这种混合物在较高温度下聚合成十氢萘产物2.1的单一区域异构体。在ζ消除之前观察到一个非选择性的β-消除途径,给出单一的十氢萘区域异构体,表明区域收敛是在发挥作用。为了证实这一观察结果,将异构体1-,2-和3-(2-皮考基)环癸烷-1-醇(分别为1.2,1.3和1.4)进行跨环ζ消除(Fig. 2C)。在所有情况下,decalins (2.2a-2.2c)的产率和比例相当,证实了假设的区域收敛性,并在syn-1.2和anti-1.2的情况下建立了立体收敛性。由于合成1.2、1.3和1.4的难易程度差异很大(1-取代1.4可以一步获得),对于所提议的底物的合成完全失败的情况,该方法的区域收敛性提供了使用底物的替代异构体作为后备。
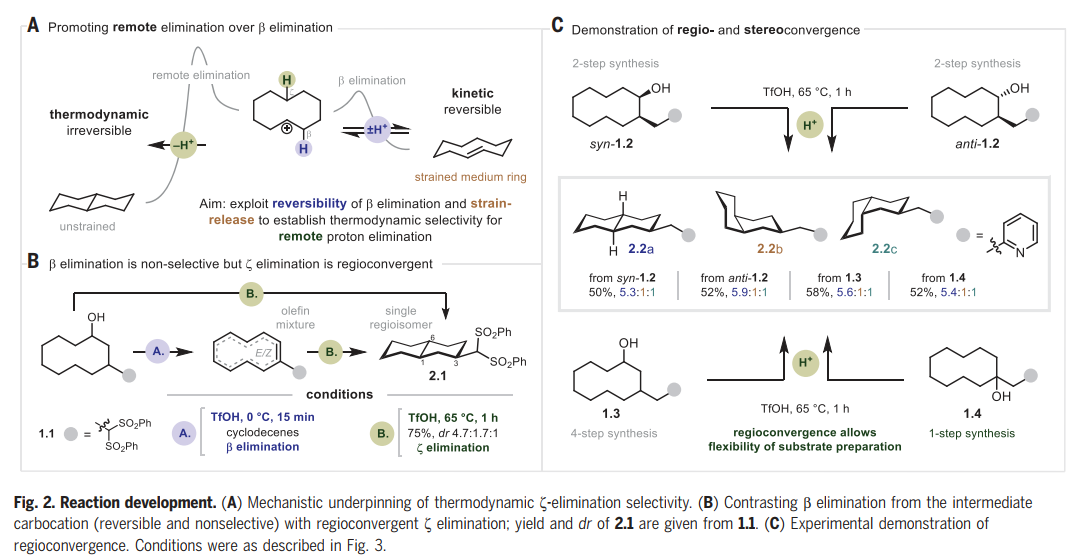
图片来源:Science
在这一点上,作者试图彻底研究几种1-、2-和3-取代环癸醇以及烯烃底物的远程消除底物范围(Fig. 3)。1-取代环癸醇(1)可以用最少的合成步骤制备,因此构成了本文研究的大部分。作者发现底物上的缺电子和富电子杂环取代基适合跨环消除,可获得吡啶取代(2.2,2.6)、喹啉取代(2.3,2.5)、吲哚取代(2.4)、苯并咪唑取代(2.7和2.15)和苯并噻唑取代(2.11)十氢萘,产率优良。在研究早期,作者试图建立该反应的合成可行性,以克为单位制备喹啉2.3,采用较温和的条件(1.5当量的TfOH),并以84%的收率提供克级规模2.3。
氨基醇和烯烃也能兼容,提供2.9,2.10,2.12,2.14和2.16至2.20。使用2-取代环癸醇可以以良好的非对映选择性高效制备2.21至2.27。最后,作者研究了3-取代环癸醇,得到2.28到2.30。该反应能同时形成三个立体中心,从而使起始原料的取代模式和相对立体化学与产物分离。在其他双环异构体中,如5,7-稠合环系,观察到十氢萘具有完全的选择性。
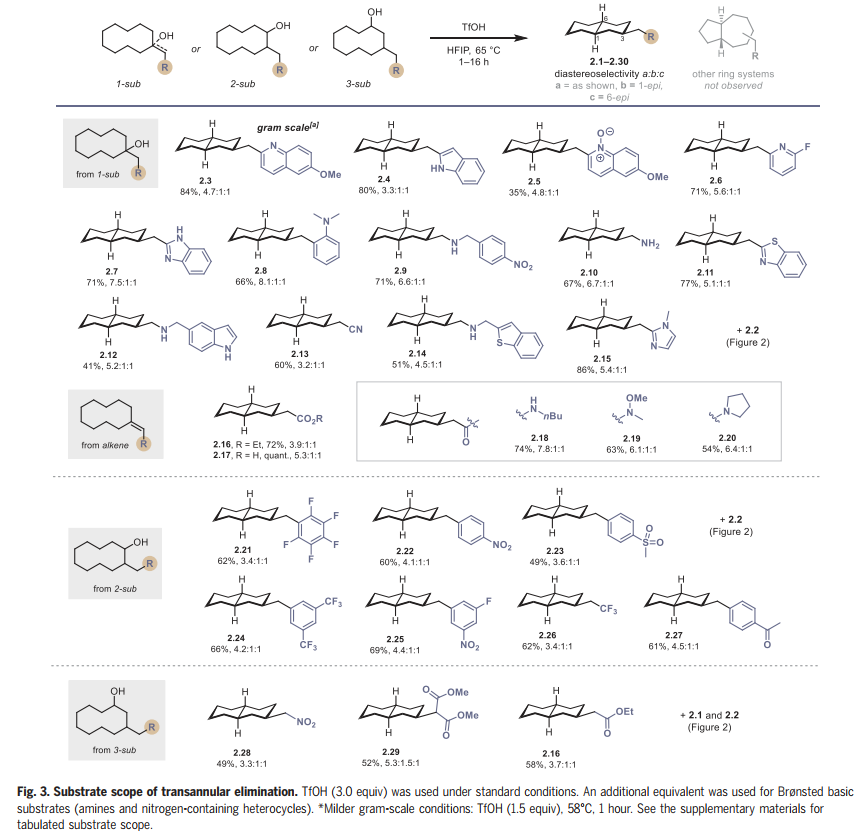
图片来源:Science
接着,作者进行了反应机理的研究。作者对环癸基阳离子在三氟甲磺酸酸盐(-OTf, A)或六氟锑酸盐(SbF6-,Sb-A)抗衡离子存在下的反应性进行了DFT计算(Fig. 4C)。在三氟甲磺酸盐存在时观察到H+的消除(产生十氢萘C-a)和在SbF6-存在时排出 (产生十氢萘阳离子Sb-C),两者之间存在明显的差异。结合实验证据,可得出结论:远端酸化是C-H活化的模式,支持观察到的反应活性,抗衡离子在介导该反应中的关键作用。此外,DFT计算使作者能够确定动力学选择性是远程消除的非对映选择性和区域选择性的主要决定因素。
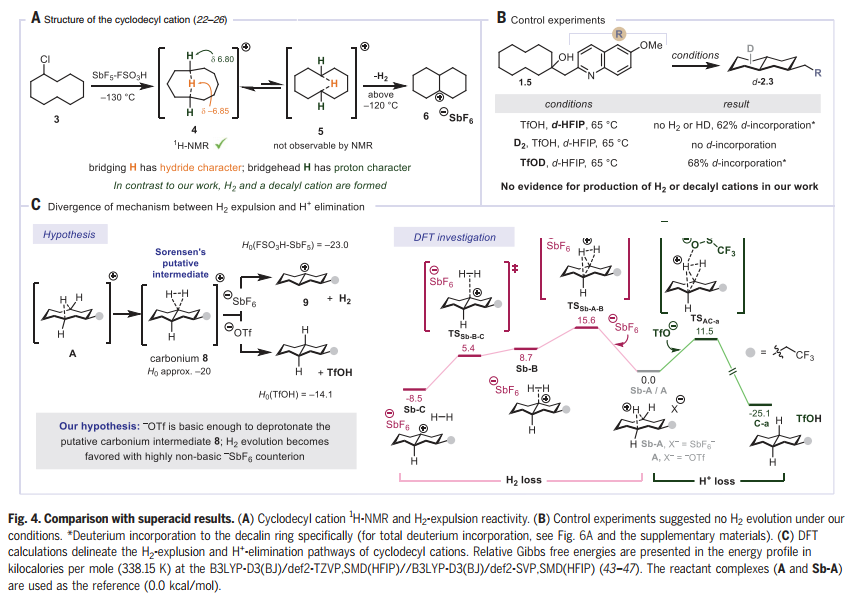
图片来源:Science
在本文的条件下,作者使用d-HFIP和TfOD,获得了全氘代十氢萘,为合成氘代十氢萘提供了一种简单而可靠的替代方案。最后,受陈弓教授等人的工作的启发,作者研究了HAT介导的远端C-H活化产物的差向异构化(Fig. 6B)。在BIN3, 14条件下,2.Xb顺利发生差向异构化,得到热力学稳定的异构体 2.Xa。
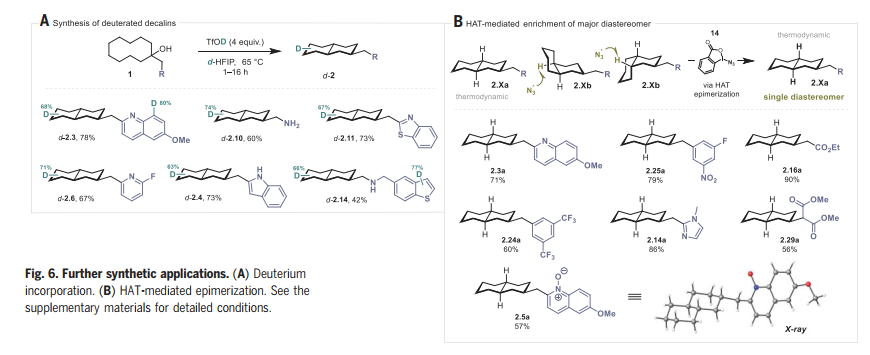
图片来源:Science
Nuno Maulide教授课题组提出了一种通过远端质子消除形成C-C σ键的方法,这是一种通过远端酸化实现的C-H活化的独特模式。将这一概念应用于环癸基阳离子,开发了一种吸引人的合成十氢萘的方法。这种转化是区域收敛的,不需要导向基或贵金属,具有精准的位点选择性,并涉及介质环的张力释放。
文献 1 详情:
Enantioselective remote methylene C-H (hetero)arylation of cycloalkane carboxylic acids.
Tao Zhang, Zi-Yu Zhang, Guowei Kang, Tao Sheng, Jie-Lun Yan, Yuan-Bin Yang,
Yuxin Ouyang, Jin-Quan Yu*.
Science, 2024
https://www.science.org/doi/10.1126/science.ado1246
文献 2 详情:
Remote proton elimination: C-H activation enabled by distal acidification.
Phillip S. Grant, Miloš Vavrík, Vincent Porte, Ricardo Meyrelles, Nuno Maulide*.
Science, 2024
https://www.science.org/doi/10.1126/science.adi8997
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国