

导读
近日,美国威斯康星大学麦迪逊分校的Shannon S. Stahl课题组报道了一种铜/次硝酸(nitroxyl)共催化的方法,在水溶剂中通过有氧脱氢和氧化偶联可将环状仲胺直接转化为相应的内酰胺衍生物。同时,该反应具有广泛的底物范围以及良好的官能团兼容性等特点。此外,通过对砌块以及复杂分子的选择性官能团化,如溴结构域抑制剂(bromodomain inhibitors)的后期官能团化,进一步证明了反应的实用性。
(图片来源:J. Am. Chem. Soc.)
正文
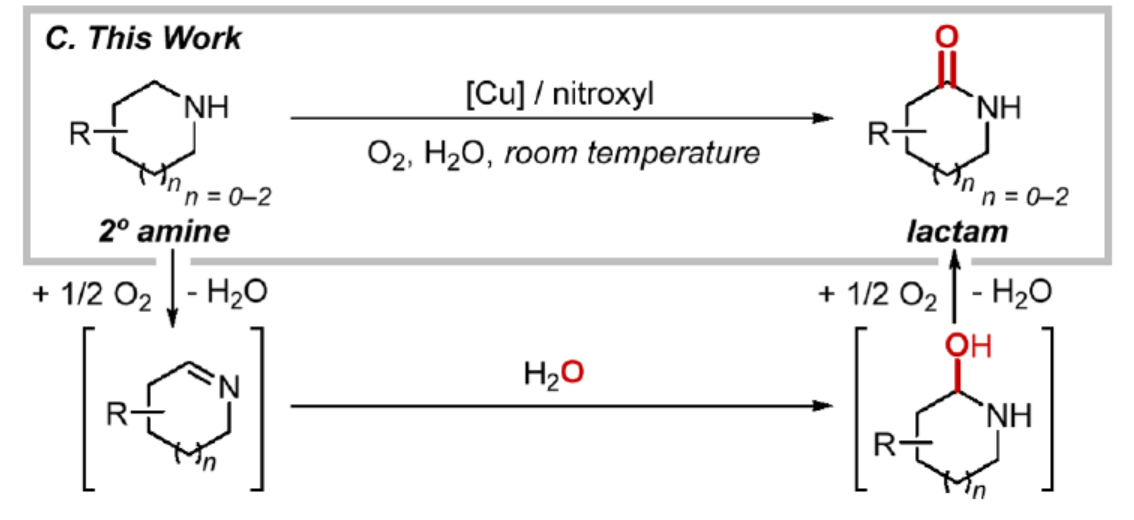
哌啶、吡咯烷和相关的饱和氮杂环因其良好的物理化学和药代动力学特性而普遍存在于药物分子中。因此,这些结构在用于药物发现的合成砌块中具有显著特征。其中,对于N-杂环衍生化的合成方法,通常利用与氮原子相邻的立体电子活化的C(sp3)−H键,并在此位置引入新的基团。然而,大多数的方法常需使用具有N-取代基或保护基(氨基甲酸酯、酰基、烷基、芳基)的底物。此外,二级(N−H)衍生物及其相应的铵盐特别丰富,但对于二级胺的官能团化却较少有相关的研究报道。同时,在N−H基团旁边引入一个氧原子,可将环胺转化为2°内酰胺,此过程极具吸引力。并且,这种转化是合成生物活性内酰胺和药物代谢产物的有效途径(Scheme 1A)。与其他α-官能团化方法一样,氧化反应通常需要具有完全取代的氮原子底物(Scheme 1B)。作为一个显著的例外,Milstein课题组(J. Am. Chem. Soc. 2014, 136, 2998.)报道了钌催化无受体脱氢方法,但由于苛刻的反应条件导致其官能团兼容性较差。近日,美国威斯康星大学麦迪逊分校的Shannon S. Stahl课题组报道了一种温和的铜/次硝酸催化的有氧氧化方法,可用于环状二级胺的α-氧化反应,合成了一系列内酰胺衍生物(Scheme 1C),并可用于一系列砌块和复杂生物活性分子的后期衍生化。
(图片来源:J. Am. Chem. Soc.)
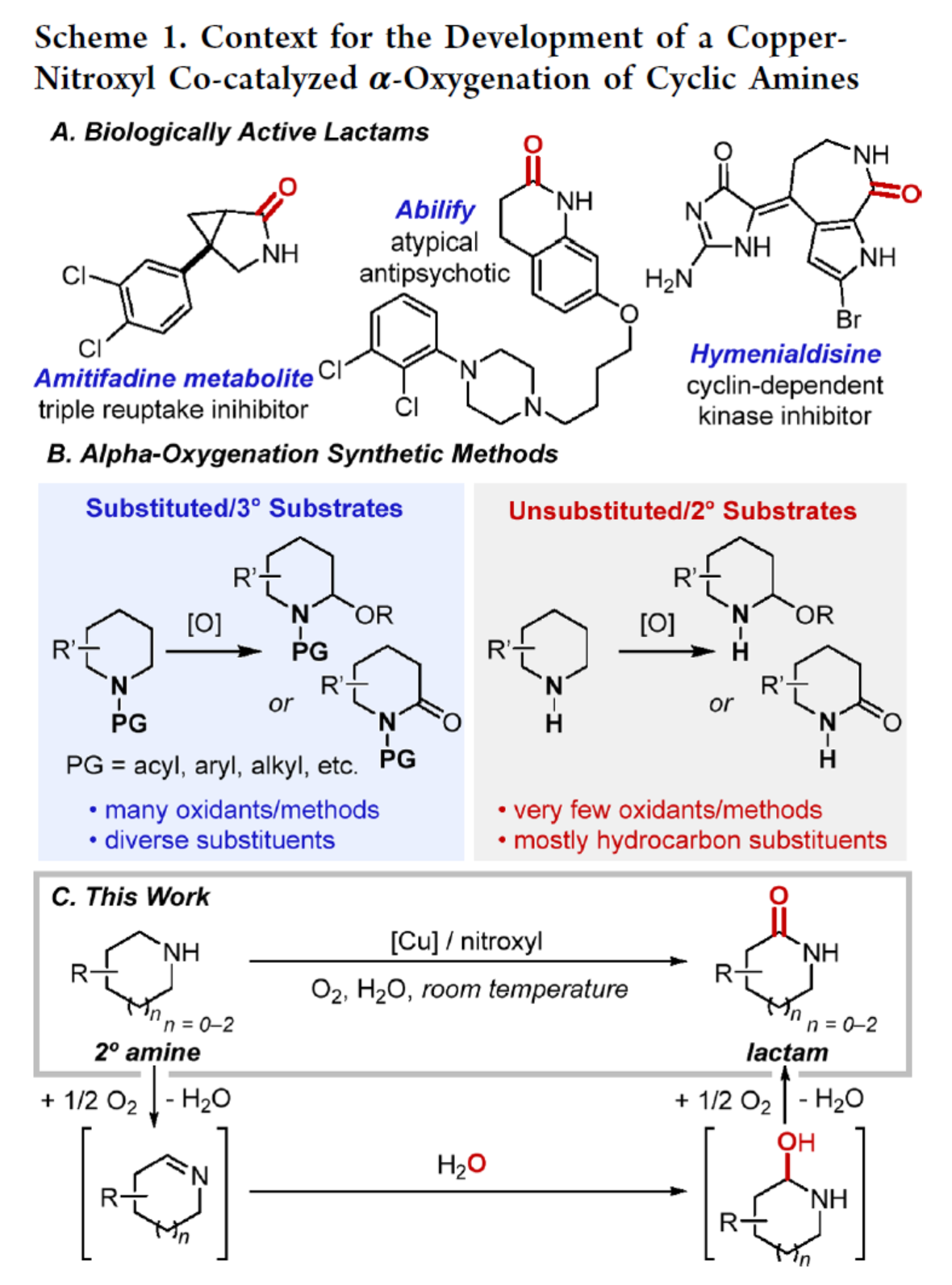
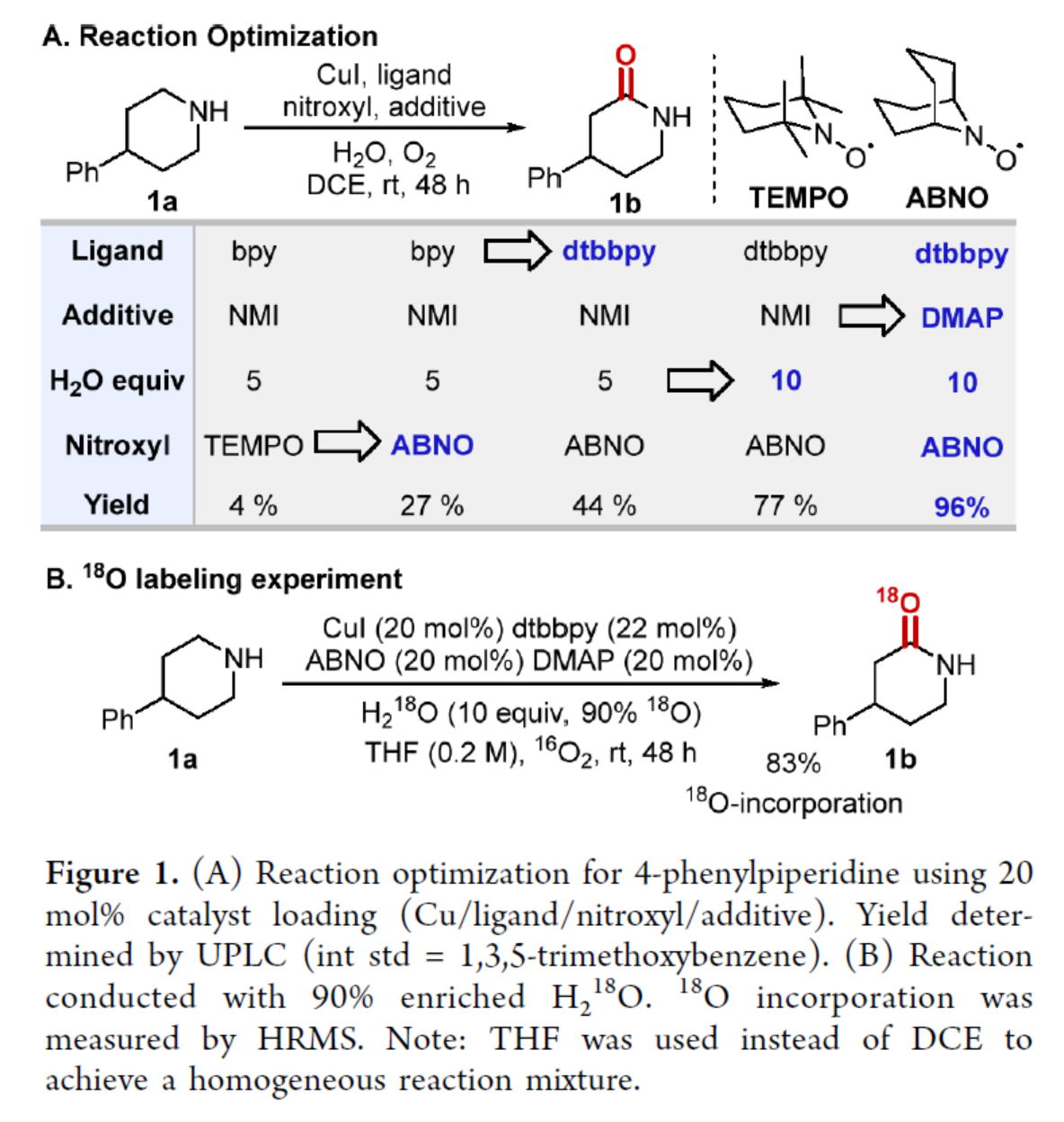
首先,作者以4-苯基哌啶1a作为模型底物,进行了相关氧化反应条件的筛选(Figure 1A)。当以CuI(20 mol %)作为催化剂,dtbbpy(22 mol %)作为配体,DMAP(20 mol %)作为添加剂,ABNO(azabicyclo[3.3.1]nonane-N-oxyl)(20 mol %)作为亚硝酰基源,氧气作为氧化剂,在水与DCE的混合溶剂中室温反应48 h,可以96%的收率得到产物1b。同时,18O氘标记实验结果表明,产物中的氧源来源于水,而非氧气(Figure 1B)。
(图片来源:J. Am. Chem. Soc.)
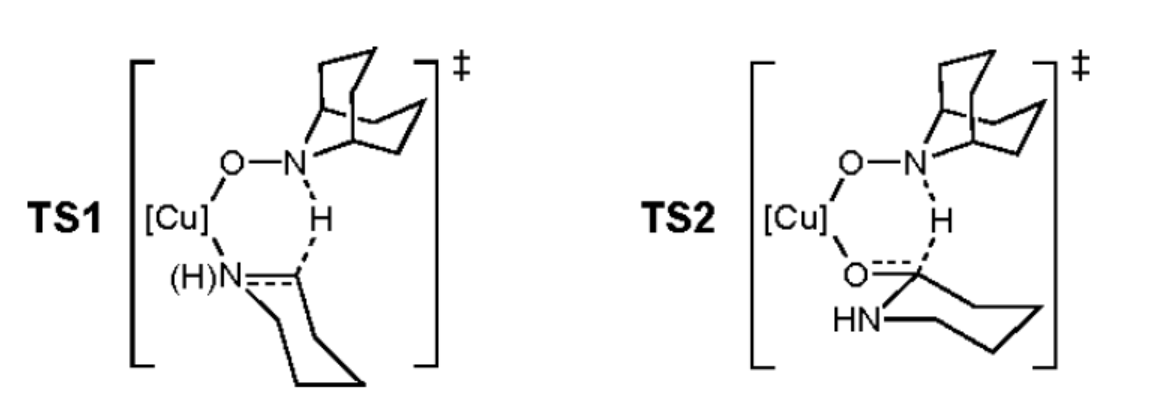
作者认为,该反应通过三元Cu/ANBO/底物复合物过渡态TS1和TS2进行,类似于醇氧化反应。同时,醇氧化比胺氧化进行得更快。因此,相对于由TS1形成亚胺中间体而言,由TS2将半胺缩醛氧化为内酰胺似乎是快速的。
(图片来源:J. Am. Chem. Soc.)
其次,通过t-分布邻域嵌入(t-SNE)分析药物相关胺砌块的分布,用于在有氧α-氧化反应条件下选择不同的底物进行评估(Figure 2A)。同时,使用六种不同的反应条件进行高通量实验(HTE),包括两种不同的CuI源(CuI和[Cu(MeCN)4]PF6)和三种不同的溶剂(DCE,MeCN和THF)(Figure 2B)。研究结果表明,反应的最佳条件随着底物的不同而发生变化。
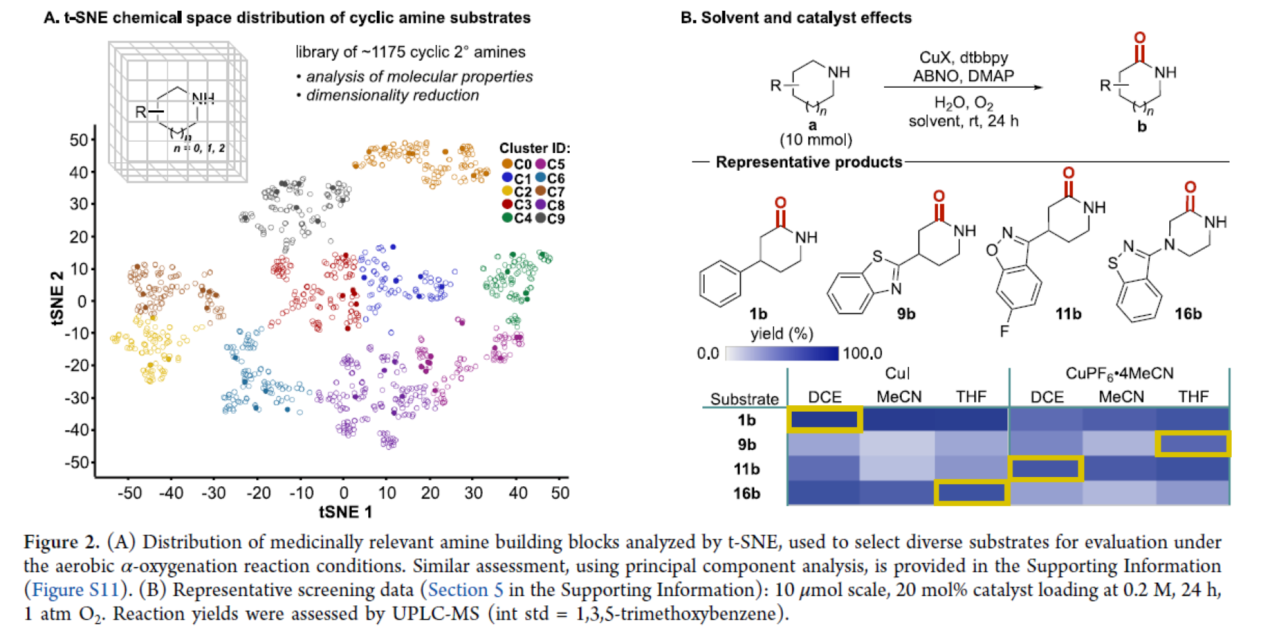
(图片来源:J. Am. Chem. Soc.)
在获得上述最佳反应条件后,作者对六元脂肪族环胺的底物范围进行了扩展(Figure 3)。研究表明,一系列不同取代的哌啶、哌嗪和吗啉衍生物,均可顺利进行反应,获得相应的产物1b-24b,收率为21-98%。其中,对于存在两个相互竞争的氧化位点的底物(如10b、13b和24b),反应优先在空间位阻较小的位点进行。同时,含有位阻较大的α-取代胺(3b、4b、7b和14b)反应效率较低。值得注意的是,在反应条件下可耐受各种官能团,包括芳基、芳基卤、酰胺、3°胺、3°醇、烷氧羰基、砜和杂芳基衍生物。

(图片来源:J. Am. Chem. Soc.)
紧接着,作者对五与七元脂肪族环胺的底物范围进行了扩展(Figure 4)。首先,一系列不同取代的吡咯烷衍生物,均可顺利进行反应,获得相应的产物25b-32b,收率为20-99%。其中,手性吡咯烷衍生物,在获得内酰胺产物(29b、30b和31b)时,能够在远程位点保留立体化学。其次,四种不同的氮杂环庚烷衍生物,也与体系兼容,获得相应的产物33b-36b,收率为46-82%。
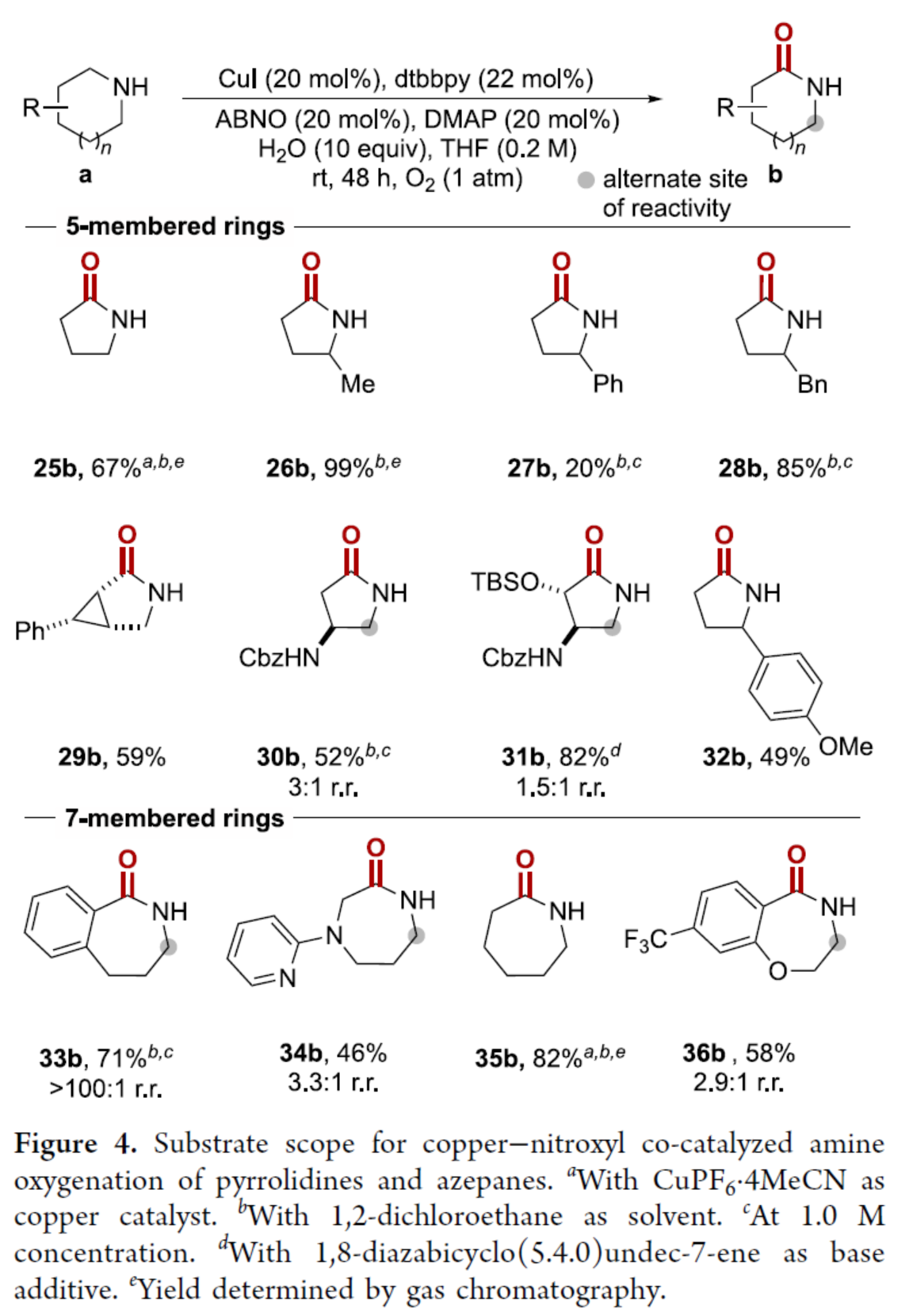
(图片来源:J. Am. Chem. Soc.)
通过对反应条件的再次优化后发现,当以胺盐为底物,通过两步一锅反应(游离碱/氧化),可直接合成内酰胺产物1b、37b和38b,收率为43-84%(Figure 5)。值得注意的是,在人类口服伐伦克林药物后,内酰胺产物38b作为主要代谢产物,进一步证明了该反应在合成代谢物合成中具有广泛的应用。
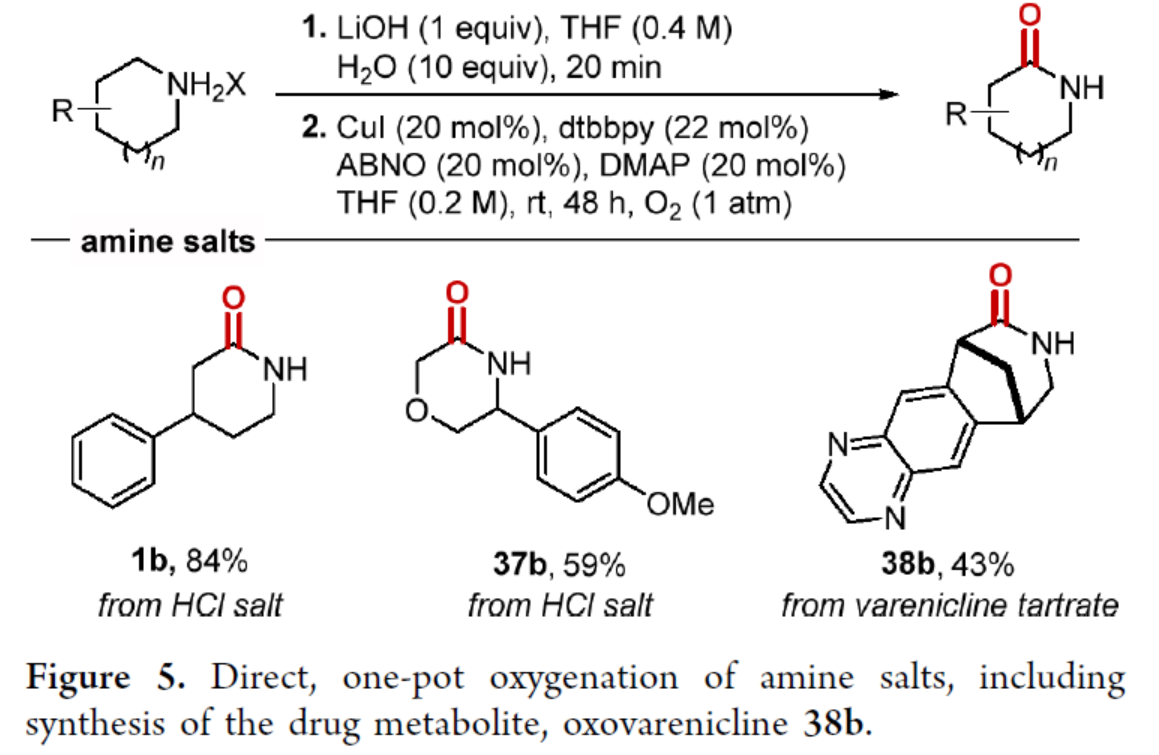
(图片来源:J. Am. Chem. Soc.)
此外,作者还对反应的实用性进行了的研究(Figure 6)。首先,溴结构域蛋白(bromodomain proteins)抑制剂是肿瘤学和免疫炎症应用中积极开发的焦点。将BET-抑制剂和BRPF1-抑制剂置于Cu/ABNO催化条件下,可以良好的收率得到区域异构体混合物39b和40b。其中,39b的单个区域异构体分别以32%和27%的收率分离出来。其次,p38激酶抑制剂也成功进行了氧化反应,可以50%的收率得到产物41b。上述实验结果进一步证明了,该方法在复杂候选药物的后期修饰的能力。

(图片来源:J. Am. Chem. Soc.)
总结
美国威斯康星大学麦迪逊分校的Shannon S. Stahl课题组开发了一种操作简单的催化方法,用于环状2°胺的α-官能团化反应,并以化学信息学为指导选择底物,以展示其在化学空间中的实用性。同时,该反应具有反应条件温和、底物范围广泛以及良好的官能团兼容性等特点。此外,通过对药物代谢产物合成以及候选药物后期官能团化,进一步证明了反应的实用性。
文献详情:
Copper−Nitroxyl-Catalyzed α‑Oxygenation of Cyclic Secondary Amines Including Application to Late-Stage Functionalization.
Christopher M. Hanneman, Jack Twilton, Melissa N. Hall, Nicole C. Goodwin, Jennifer M. Elward, Tessa Lynch-Colameta, Shannon S. Stahl.J. Am. Chem. Soc. 2024https://doi.org/10.1021/jacs.4c04359



 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国