

导读
近日,美国芝加哥大学(University of Chicago)董广彬课题组和美国匹兹堡大学(University of Pittsburgh)刘鹏课题组通过金属络合物中可能的张力释放机理加速协同的SNV反应,从而实现了有机硼酸酯的通用和立体特异性的亚乙烯基同系化反应。该方法使多个亚乙烯基单元的迭代结合成为可能,从而得到具有合成挑战性的交叉共轭多烯类化合物。此外,作者通过产物的进一步合成转化实现了含有多取代烯烃的生物活性化合物的合成,证明了此转化的实用性。计算研究表明,在方形平面过渡态中,空间张力的减小促进了一种不同寻常的SN2类型的协同反应路径,由此解释了该金属SNV反应的高效和立体反转特性。相关成果发表在Nature上,文章链接DOI:10.1038/s41586-024-07579-7。
(图片来源:Nature)
正文
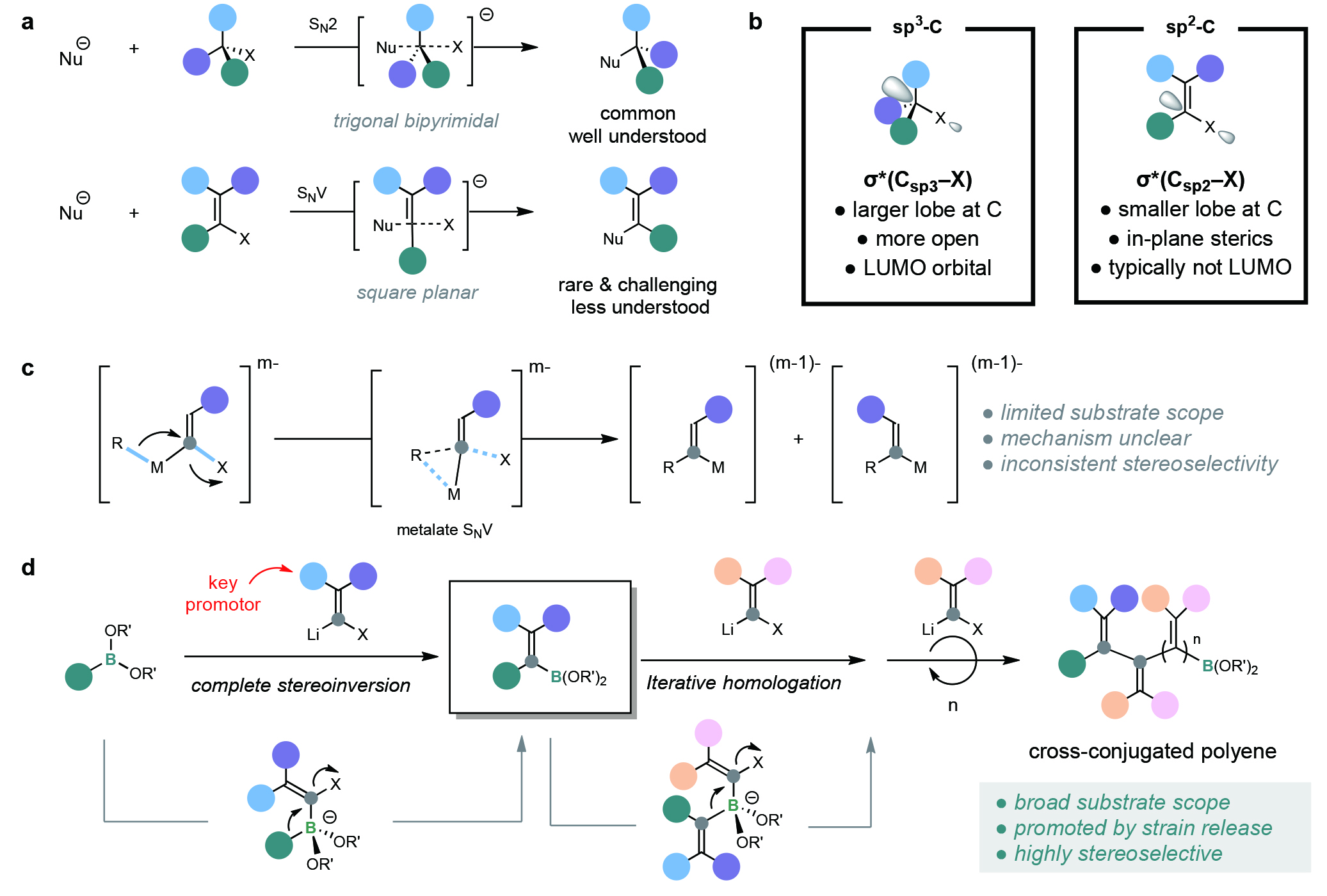
协同亲核取代反应,又被称为SN2反应,是有机合成中引入新的官能团和构建碳-碳和碳-杂原子键的基本有机转化。SN2反应通常涉及亲核试剂对C(sp3)−X键(X = 卤素或其他离去基团)σ*轨道的反向进攻,从而导致立体中心的完全反转。相比之下,在sp2乙烯基亲电试剂上进行相应的立体反转的亲核取代,即协同SNV(σ)反应是非常罕见的,到目前为止,仅限于机理精心设计的底物可以实现此过程,且主要是在环形成过程中实现的。最近,美国芝加哥大学董广彬课题组和美国匹兹堡大学刘鹏课题组通过金属络合物中的张力释放机理加速协同的SNV反应,从而实现了有机硼酸酯的通用和立体特异性的亚乙烯基同系化反应(Fig 1)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
(图片来源:Nature)
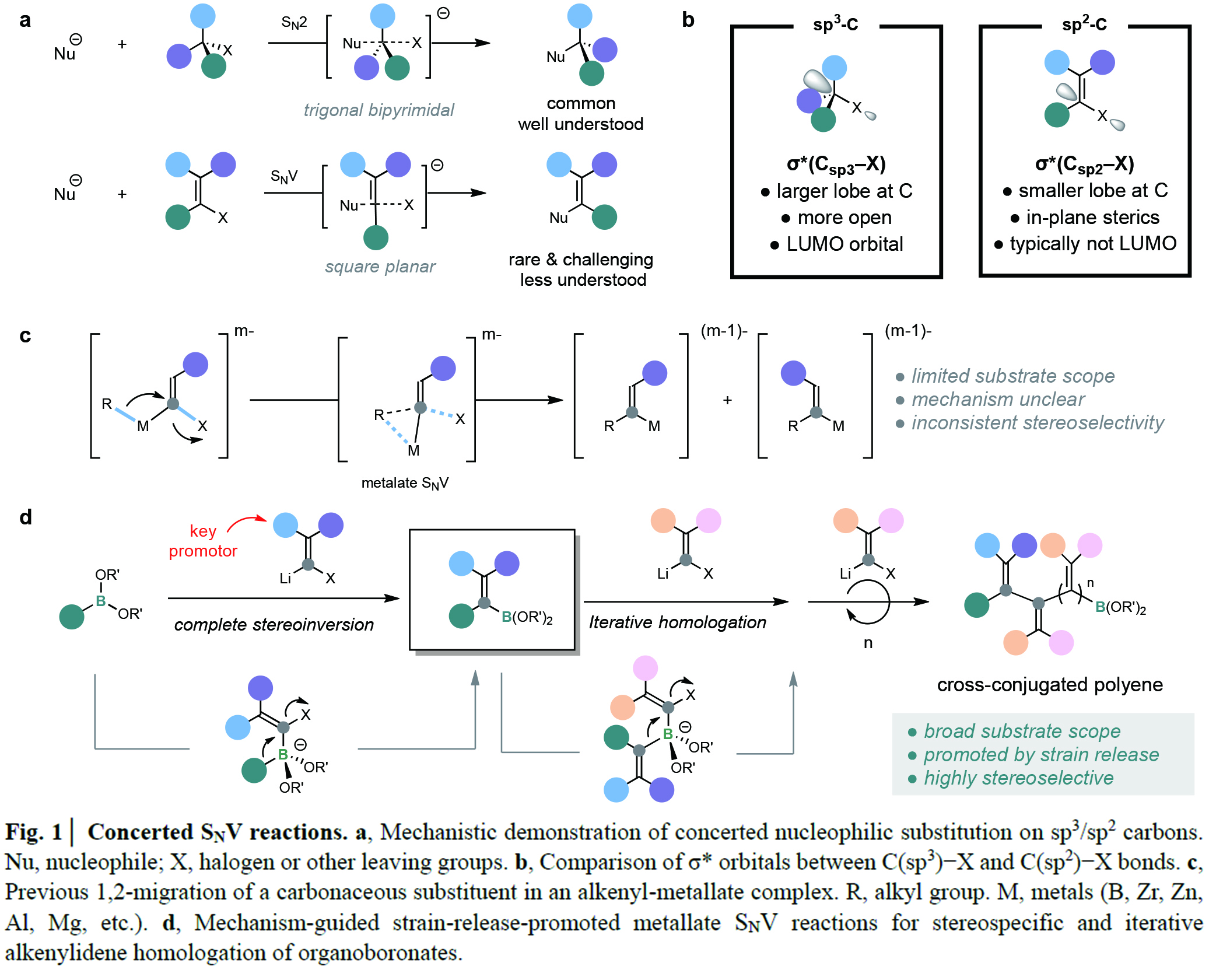
首先,作者以芳基硼酸酯1a-Tgly, 烯基溴试剂E-2b作为模板底物对反应进行了探索,并通过条件优化得出最优反应条件为:1a-Tgly (1.0 equiv.), E-2b (1.5 equiv.), LiBr (1.0 equiv.), LiTMP (1.5 equiv.),在乙醚(0.1 M)中,-78 oC反应可以以89%的产率得到产物Z-3b’。此外,DFT计算表明该反应经历了一个协同的1,2-迁移过程,由于硼酸酯的氧与顺式的苯取代基存在空间排斥,因此会通过平面过渡态E-TS-2a的空间张力释放以及基团的迁移实现与苯取代基的π-π相互作用,因此实现了位阻更大的E-式烯烃的构型反转(Fig 2)。
(图片来源:Nature)
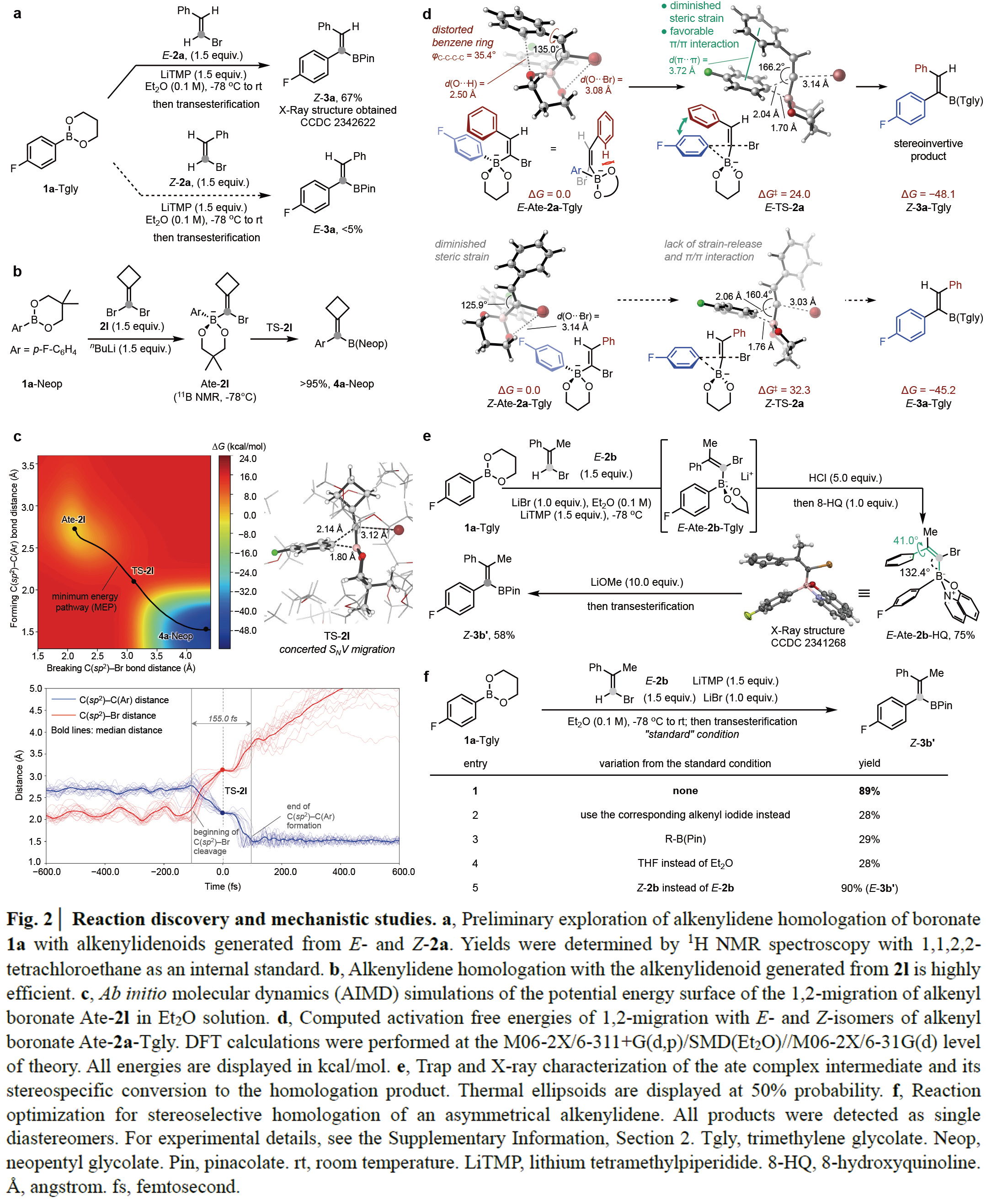
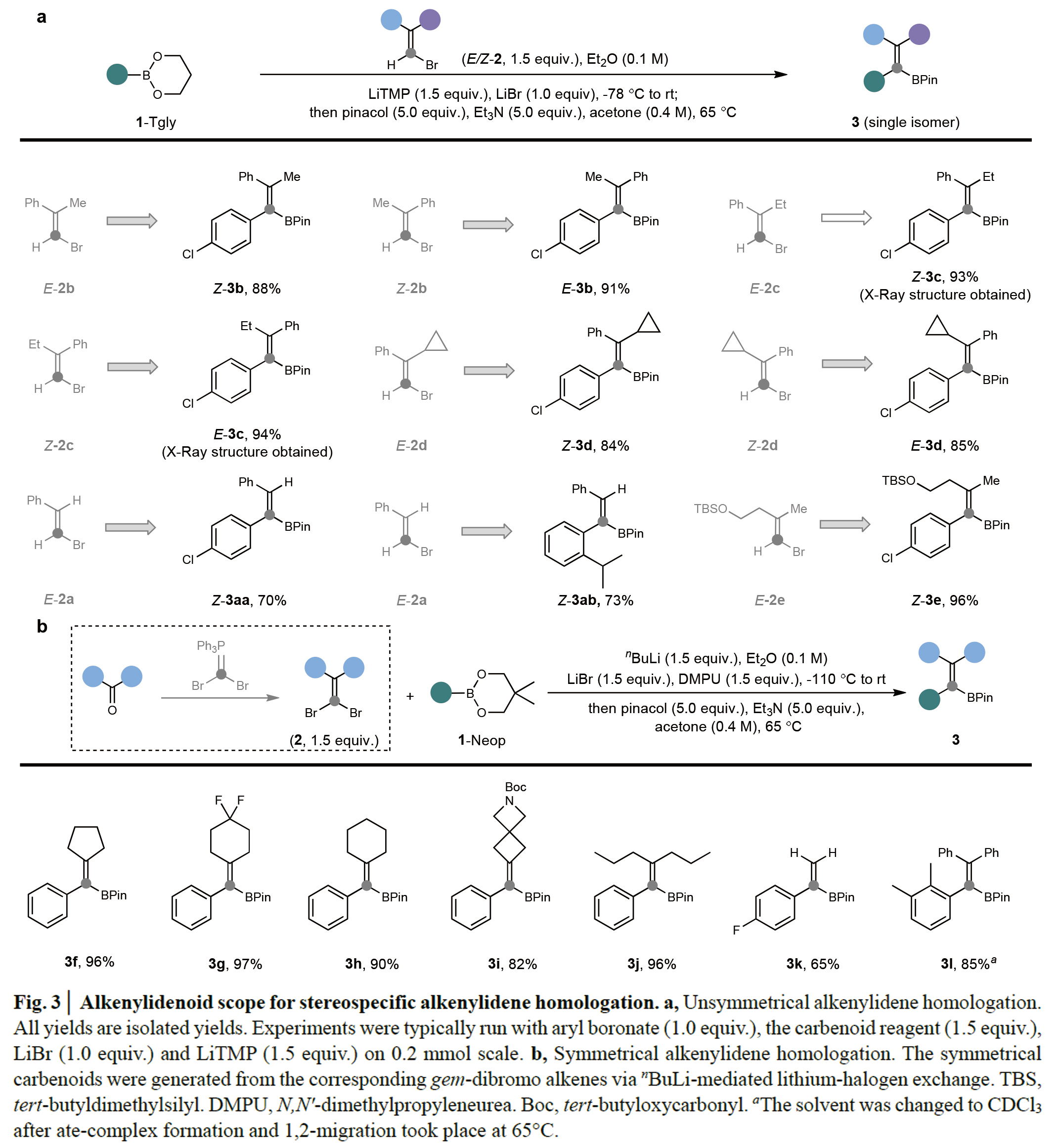
在得到了最优反应条件后,作者首先对一系列非对称的烯基溴试剂的适用范围进行了考察(Fig 3)。首先,无论2,2-二取代烯基溴(2b-2d)的E-和Z-异构体均可以经过α-去质子化和同系化过程,以84-94%的产率,以单一的几何异构体得到相应的烯基硼酸酯产物(其它异构体未被粗反应混合物的核磁共振检测到),这样就可以以高效的立体选择性得到非对称的四取代烯烃。该反应的高立体特异性不仅支持了反应涉及SNV反应途径,还表明了不希望的金属辅助异构化过程和α-消除形成游离卡宾的速度明显慢于ate-络合物的形成。此外,可以很容易从相应的酮来获得的结构多样的对称烯基溴试剂也可以顺利实现转化,以65-96%的产率得到所需产物3f-3l。Tgly和Neop硼酸酯与对称的烯基溴试剂均可反应,且Neop硼酸酯由于更容易纯化操作而可以广泛被使用。与二取代烯基溴试剂的优良反应性相比,简单的烯基溴试剂也可以顺利实现亚乙烯基部分的插入(3k),但产率较低(65%)。
(图片来源:Nature)
令人欣慰的是,硼酸酯在亚烯基同系化反应中同样展现出广泛的范围(Fig 4)。具有不同电性的的芳基硼酸酯均可顺利实现转化,以56-99%的产率得到相应的产物4a-4z, 4aa-4an。其中叔胺(4o)、酯(4q)、噻吩(4s)、呋喃(4t)、烯烃(4v)和炔(4aa)等多种官能团均可兼容。此外,作者还考察了迁移芳基的立体效应。一般来说,具有对位取代基、间位取代基和邻位取代基的芳基的底物均可以以良好的产率得到产物。值得注意的是,大位阻的邻异丙基苯基(4ag)和均三甲苯(4al)硼酸酯同样是合适的底物,这意味着在此转化中可以兼容大空间位阻取代基。除了芳基硼酸酯以外,烯基硼酸酯也是合适的底物(4ab)。此外,一级硼酸酯和二级硼酸酯,以及具有不同环尺寸(5i-5l)的环硼酸酯均可顺利实现同系化。反应中的官能团,如烷基溴(5e)、叠氮(5f)和端烯(5g)均在反应中保持不变。
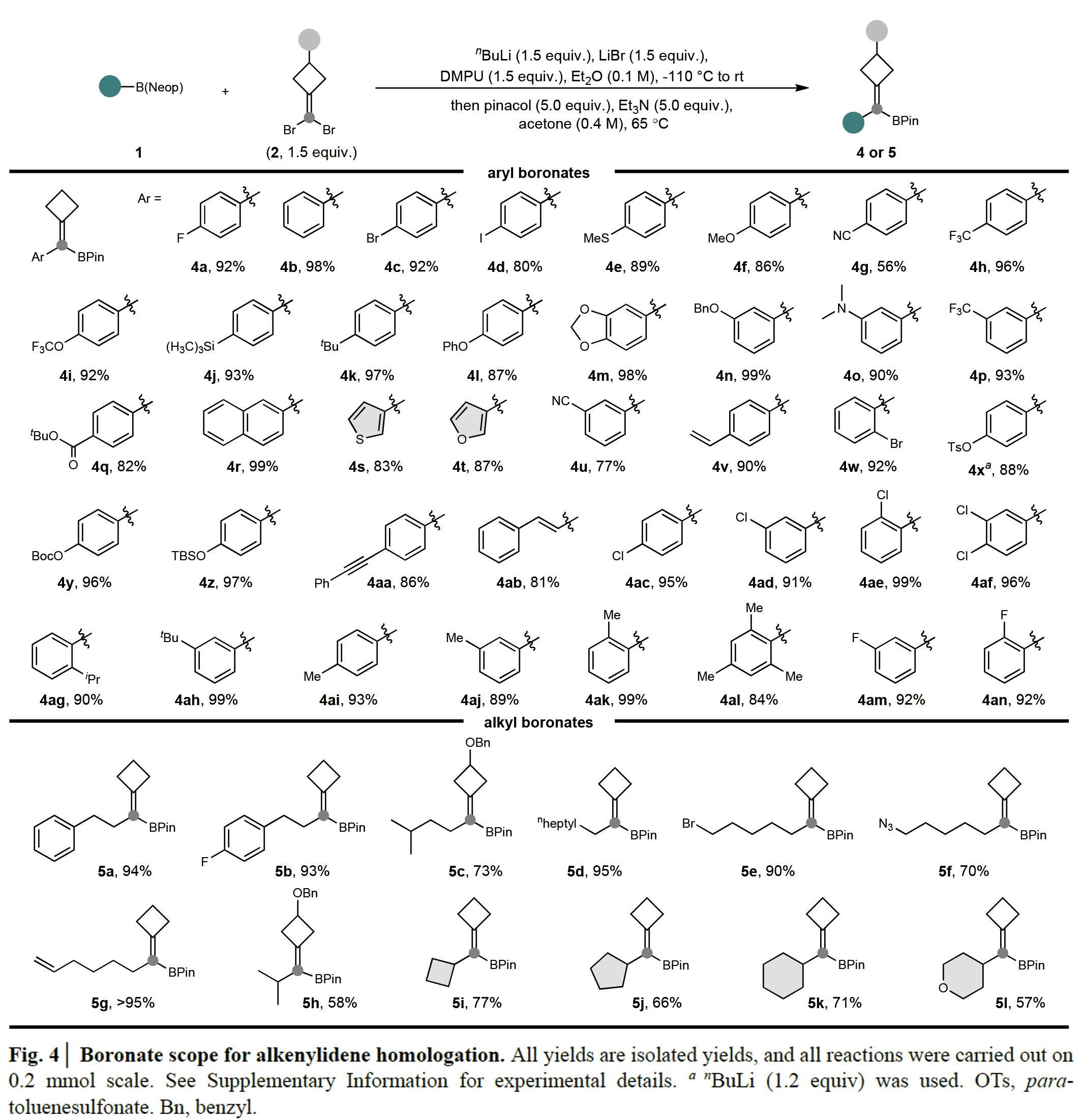
(图片来源:Nature)
接下来,作者利用此迭代同系化过程来制备通常难以合成的共轭多烯(Fig 5)。从4-氟苯基新硼酸酯(1a-Neop)开始,利用一锅双同系化可以以85%的产率得到二烯硼酸酯6a。且将粗样品通过硅胶过滤去除无机盐和大极性副产物后,通过进一步的同系化还可以实现链的延伸。作者采用相同的迭代顺序,分别成功合成了三烯(6b,71%)、四烯(6c,57%)和五烯(6d,45%),且整体效率较高。值得注意的是,所有的烯烃都是四取代的,可以想象间隔的环丁基取代基之间的具有较大的立体排斥作用。值得注意的是,完全取代的交叉共轭四烯和五烯目前还尚未有报道。不同种类的亚乙烯基砌块可以以可编程的方式迭代地结合以构建结构不同的交叉共轭多烯(6e-6g)。此外,作者还合成了一系列通过两个非对称亚乙烯基单元依次插入所得到的交叉共轭二烯(6h-6k)。由于该方法避免了高温条件和过渡金属的使用,因此作者并未观察到这些交叉共轭多烯的异构化过程发生。
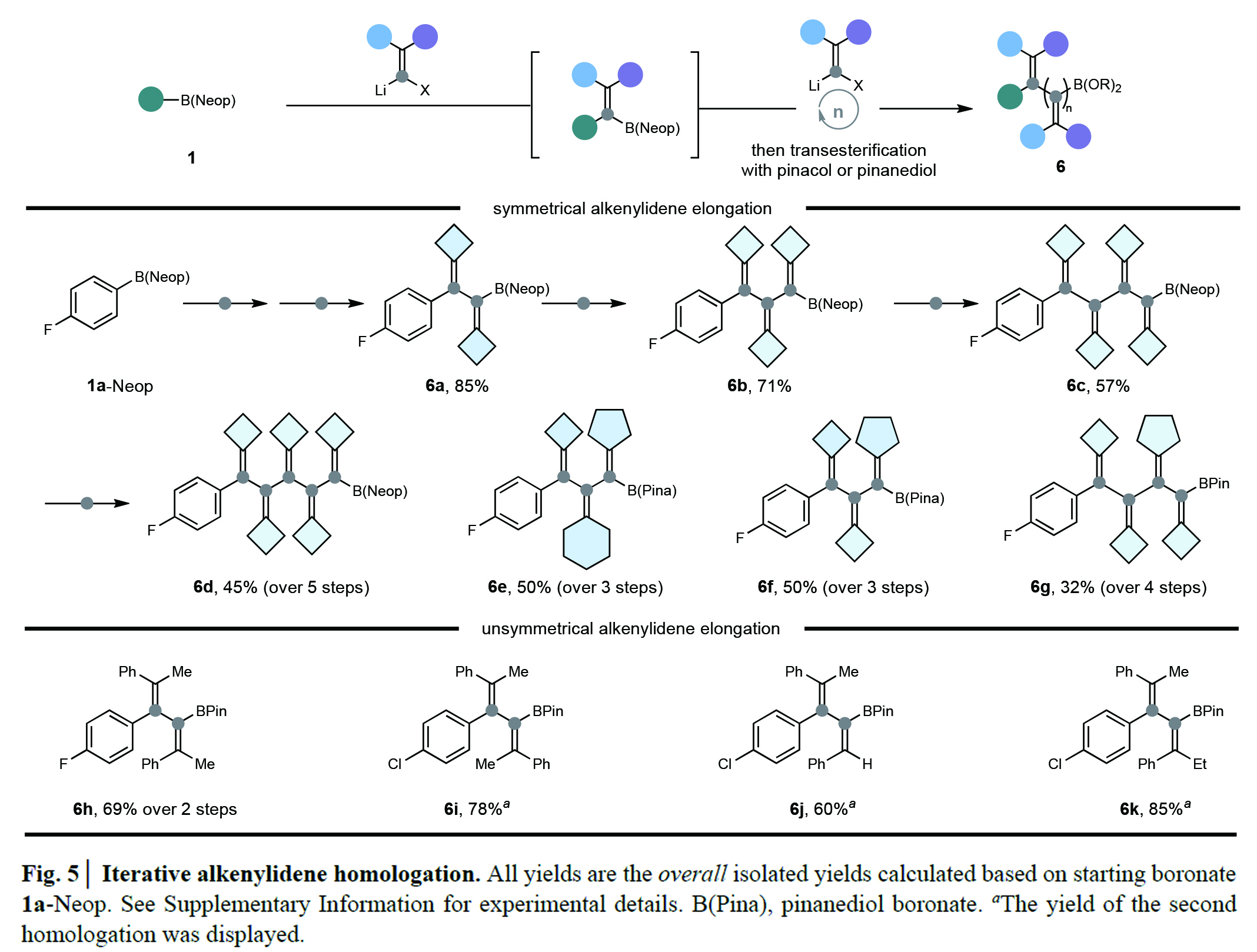
(图片来源:Nature)
多取代烯烃广泛存在于生物活性分子和药物中。因此,将亚乙烯基同系化和原位Suzuki偶联串联可以实现多取代烯烃的模块化、快速和立体特异性制备(Extended Data Fig. 1)。例如,4-乙烯基哌啶片段通常存在于许多酶抑制剂骨架中。从偕二溴烯烃2m开始,与硼酸酯7a进行同系化,随后经历与碘苯8a的Suzuki交叉偶联反应可以得到所需的烯烃9a/9a’,总收率为93%。随后通过亲核芳香取代(SNAr)反应得到目标化合物10,该化合物是合成脂肪酸酰胺水解酶(HAAH)抑制剂的关键中间体。与先前基于逐步烯烃构造的路线相比,该路线具有更少的步骤和更少的分离纯化过程。此外,利用相同的同系化/Suzuki交叉偶联方法可以实现制备δ-阿片受体激动剂的关键中间体四取代烯烃9b(90%)和9c(85%)的合成。此外,化疗药物贝沙罗特、用于绝经妇女的候选药物GSK232802、 抗癌药物他莫昔芬及其类似物Droloxifene均可以通过此策略有效地制备。这些合成过程与已知方法相比,所有的产物均为单一的非对映体,且反应所涉及的步骤更少,总产率更高,展示了此方法的多功能性和效率。
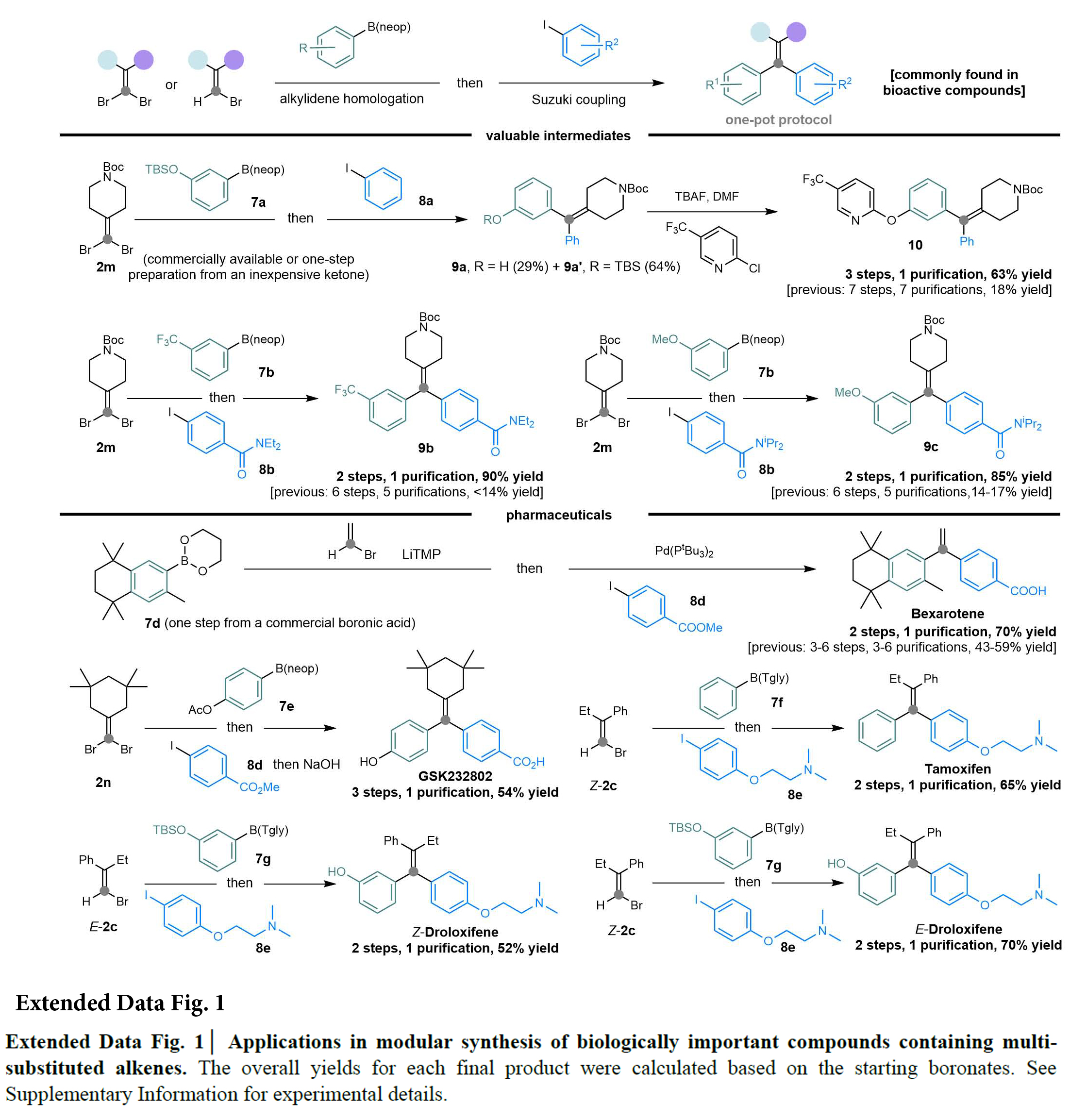
(图片来源:Nature)
总结
董广彬和美刘鹏课题组发展了一种通用的、高效的、立体特异性的亚乙烯基同系化方法,该方法是通过一种独特SNV机理实现的。该反应耐受一系列官能团,以及底物的空间位阻和电性变化。该反应的优异立体特异性使其适合于常存在于复杂的生物活性化合物中的四取代烯烃的模块化制备。此外,利用迭代的亚乙烯基同系化过程为完全取代的共轭多烯的合成提供了一条新的途径。
文献详情:
Stereospecific alkenylidene homologation of organoboronates by SNV reaction.
Miao Chen, Christian D. Knox, Mithun C.
Madhusudhanan, Thomas H. Tugwell, Coco Liu, Peng Liu, Guangbin Dong.
Nature, 2024
https://doi.org/10.1038/s41586-024-07579-7.


 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国