

导读
近日,德国哥廷根大学的Lutz Ackermann课题组报道了一种镍催化芳基锍鎓盐(arylsulfonium salts)的C(sp2)–C(sp3)交叉亲电偶联反应,成功实现了将脂肪族链引入至芳香族骨架中。因此,简单的非预官能团化芳烃可以通过形成芳基二苯并硫蒽鎓盐(DBT+,aryldibenzothiophenium salts)从而实现相应的烷基化反应。该反应采用电化学方法避免了具有潜在危险性的化学氧化还原剂的使用。重要的是,一锅法烷基化也是可行的,进一步证明了该方法的稳健性。文章链接DOI:10.1002/anie.202401198

(图片来源:Angew. Chem. Int. Ed.)
正文
将sp3-杂化碳引入至芳香族骨架上是提高药物发现临床成功率的可靠策略之一。在这种情况下,C(sp2)–C(sp3)键形成反应发挥了至关重要的作用,可合成各种三维分子。尽管与有机金属试剂的常规交叉偶联反应是高度选择性的,但常需在惰性气氛下进行。因此,化学家们已经探索了利用化学还原剂与有机卤化物进行光化学或电化学的交叉亲电偶联反应(XECs)(Scheme 1, top)。然而,这些卤化物,特别是(杂)芳基卤化物的化学空间仍然有限,因为位点选择性卤化仍然具有挑战性。为了解决这个问题,Procter、Ritter和Yolimitsu等课题组报道了相关芳基锍鎓盐的位点选择性制备和官能团化反应。2021年,Ritter课题组(J. Am. Chem. Soc. 2021, 143, 7909.)报道了一种钯催化芳基噻蒽鎓盐(TT+,arylthianthrenium)的位点选择性烷基化反应(Scheme 1,middle)。尽管取得了这些重大进展,但在镍催化下直接与芳基磺酸盐构建C(sp2)–C(sp3)键的方法仍然难以捉摸。近日,德国哥廷根大学的Lutz Ackermann课题组报道了一种镍催化芳基二苯并硫蒽鎓盐(由市售的芳烃制备)的电化学亲电交叉偶联反应(eXEC)(Scheme 1,bottom)。此外,利用电作为还原剂来避免潜在的危险化学氧化还原剂。值得注意的是,该方法提供了首次在没有纯化芳基锍鎓盐的情况下,从未经修饰的芳烃进行一锅形式C(sp2)–H烷基化的例子。化学加——科学家创业合伙人,欢迎下载化学加APP关注。

(图片来源:Angew. Chem. Int. Ed.)
首先,作者以芳基二苯并硫蒽鎓盐1与1-碘辛烷2作为模型底物,进行了相关反应条件的筛选(Table 1)。当以NiBr2(dtbbpy)(10 mol %)作为催化剂,Zn/Ni作为电极,nBu4NI作为电解质,3Å MS作为添加剂,电流为5 mA,在DMA溶剂中60 oC反应6 h,可以77%的分离收率得到产物3。

(图片来源:Angew. Chem. Int. Ed.)
在获得上述最佳反应条件后,作者分别对DBT+盐与烷基碘的底物范围进行了扩展(Figure 1)。首先,对位修饰的DBT+盐(3-8)、苯衍生的DBT+盐(9)、间-/对-位二取代DBT+盐(10-13)、邻-/间-位二取代DBT+盐(14和15)与间-/对-位三取代DBT+盐(16),均可顺利反应,获得相应的产物,收率为53-83%。同时,噻吩和喹啉衍生的DBT+盐,也是合适的底物,获得相应的产物17(收率为64%)和18(收率为43%)。有趣的是,当使用苯丙氨酸衍生物,仅在对位进行烷基化,可以40%的收率得到邻和对位异构体的混合产物19。值得注意的是,该策略还可用于一系列药物分子的后期衍生化,如氟比洛芬、美西律和布洛芬,获得相应的产物20-22,收率为45-68%。其次,具有一级脂肪链的简单碘化物,也与体系兼容,获得相应的产物23-27,收率为60-78%。类似地,含有氮或氧烷基碘化物,也是合适的底物,获得相应的产物28-30,收率为53-62%。此外,无环和环状二级烷基碘化物,也能够顺利进行反应,获得相应的产物31-37,收率为60-68%。

(图片来源:Angew. Chem. Int. Ed.)
紧接着,作者对反应的实用性进行了研究(Scheme 2)。首先,克级规模实验,同样能够以62%收率得到产物7(Scheme 2A)。其次,以非预官能团化的芳烃为底物,通过一锅烷基化反应,可获得相应的产物3、7、11、12和31,收率为40-56%(Scheme 2B)。值得注意的是,该策略作为首个利用非预官能团化的芳烃通过形成锍鎓盐进行一锅烷基化反应的例子。

(图片来源:Angew. Chem. Int. Ed.)
此外,作者还对反应机理进行了进一步的研究(Figure 2)。首先,自由基钟实验与手性烷基碘的外消旋实验结果表明,反应涉及烷基自由基的生成(Figure 2A)。其次,利用D2O捕获芳基锌配合物的反应未生成任何氘化产物,排除了芳基锌的累积(Figure 2B)。CV实验结果表明,电子更容易向芳基DBT+盐转移(Figure 2C)。基于上述的研究以及相关文献的查阅,作者提出了一种合理的催化循环过程(Figure 2D)。首先,镍(I)配合物A与芳基DBT+盐经氧化加成生成镍(III)中间体B。其次,中间体B在阴极上还原,生成芳基-镍(I)配合物C。随后,配合物C与烷基碘经单电子转移(SET)与自由基重组,生成镍(III)配合物D。最后,配合物D经还原消除,可获得目标偶联产物,并再生镍催化剂A,以完成催化循环的过程。此外,目前不能排除芳基DBT+盐直接阴极还原的机理。

(图片来源:Angew. Chem. Int. Ed.)
为了进一步证明上述的反应机理,作者还进行了相关的DFT研究(Figure 3)。计算结果表明,该反应涉及ISET(inner sphere electron transfer)、自由基重组与还原消除的过程。
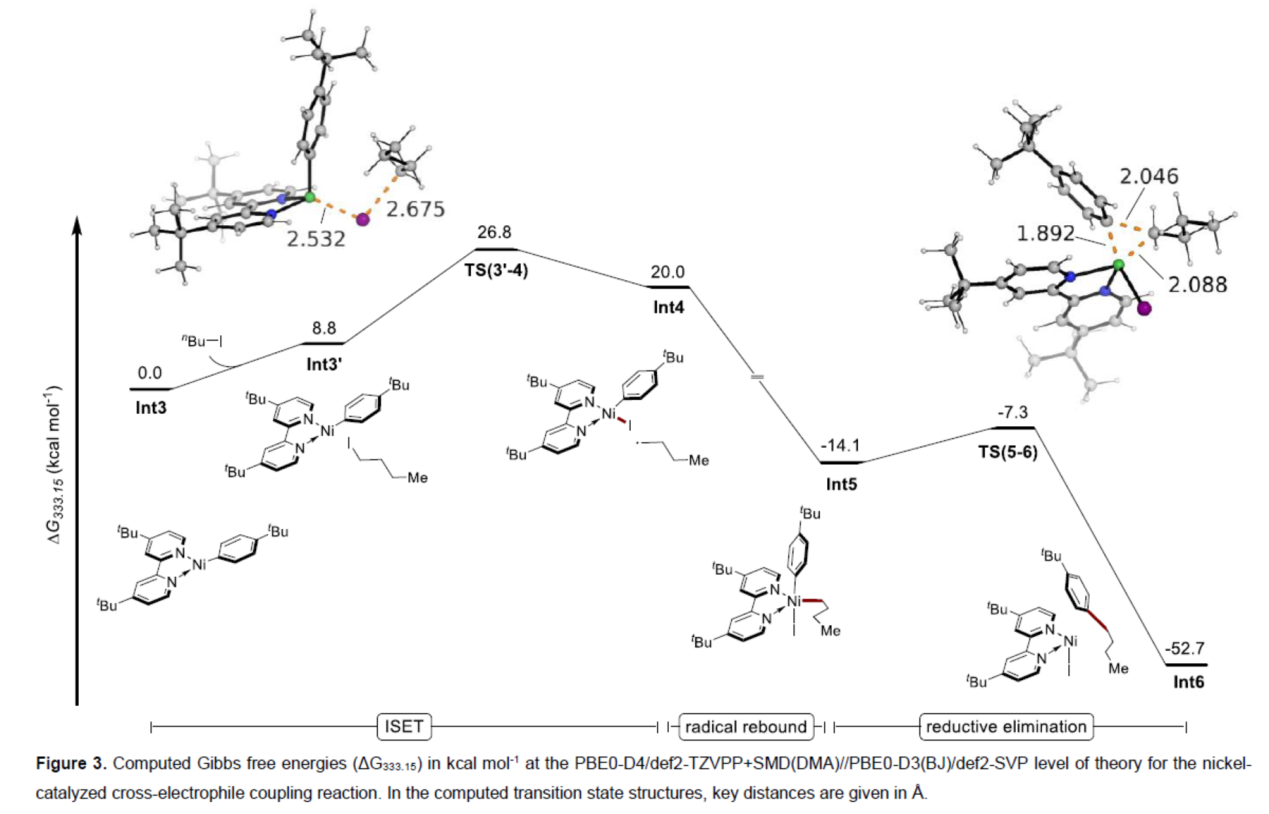
(图片来源:Angew. Chem. Int. Ed.)
总结
德国哥廷根大学的Lutz Ackermann课题组报道了一种镍-电催化C(sp2)–C(sp3) 交叉亲电偶联反应。其中,该策略可以使用各种非预官团能化的芳烃作为底物。同时,该反应具有出色的官能团耐受性与高度的化学选择性等特点。此外,一锅法电催化也是可行的,强调该方法的稳健和实用性。总的来说,该策略提供了一种有效的方法,通过未经修饰的芳烃来提高sp3-杂化的碳含量。
文献详情:
Electrocatalytic Formal C(sp2)–H Alkylations via Nickel-Catalyzed Cross-Electrophile Coupling with Versatile Arylsulfonium Salts.
Takuya Michiyuki, Simon L. Homölle, Neeraj Kumar Pandit, Lutz Ackermann*.
Angew. Chem. Int. Ed. 2024
https://doi.org/10.1002/anie.202401198
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国