

2024年5月AlphaFold 3问世,在预测生物分子结构方面再次升级,不仅提升了预测精度,其适用性也更加广泛,很多人认为这是具有里程碑意义的进展。人工智能究竟是如何预测蛋白质结构的,为什么它的准确率如此之高?本文将简要介绍AlphaFold系列预测蛋白质结构的基本原理。
撰文 | 陈清扬
神秘的蛋白质折叠
提到蛋白质,大家首先可能想到,它是一种人体必需的营养素,也可能会想到煎蛋、牛排、或是一锅美美的鲫鱼豆腐汤。蛋白质是美食的代名词,更是生命功能的物质基础,处处发挥作用:负责运送氧气的血红蛋白是蛋白质;帮助我们消化食物的消化酶是蛋白质;胰岛素、甲状腺素等激素是蛋白质;参加免疫反应的抗体也是蛋白质……据估计,人体内至少存在2万种不同类型的蛋白质,这些蛋白质的结构和功能千差万别,但构成它们的基本元素是一样的——由20种氨基酸通过不同的排列组合而构成,譬如胰岛素就是由16种、51个氨基酸构成的。
当不同的氨基酸连成一串的时候,它们会脱水形成肽链,也就是蛋白质的一级结构;而这样的一级结构是不稳定的,受到疏水作用、氢键和范德华力等影响,肽链最终会折叠成一个非常复杂而稳定的三维结构。图1是一个蛋白质折叠前后对比的例子。
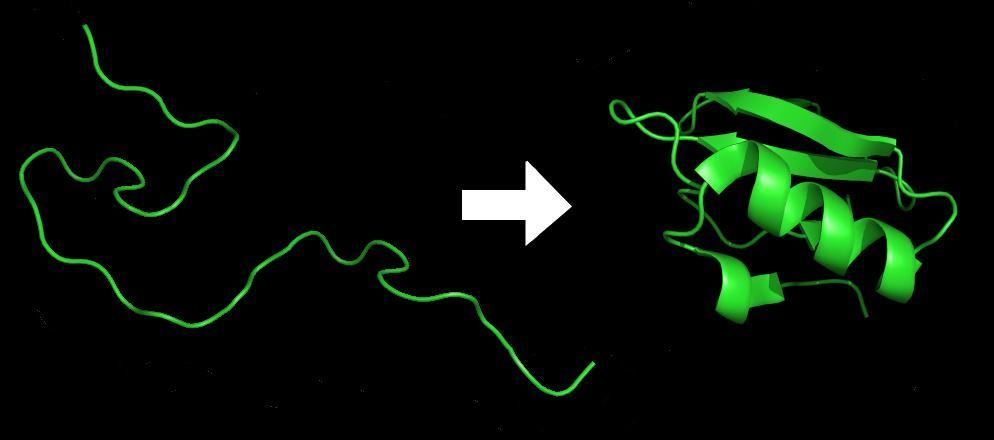
图1:蛋白质折叠示意图
蛋白质折叠成何种结构决定了它将具有何种功能,于是理解蛋白质如何折叠就成了一个十分重要的研究课题,这便是“蛋白质折叠问题”。一个蛋白质如果因为各种原因而没有正确折叠,就有可能不会正常发挥其功能,从而引发疾病,阿尔兹海默病、帕金森等疾病都和蛋白质的错误折叠有关。此外,在药物设计上面,研究人员常常需要开发具有特定功能的蛋白质,而这需要对蛋白质折叠有深入理解。
1970年代,美国生物学家、诺贝尔奖得主克里斯蒂安·安芬森(Christian B. Anfinsen)提出[1]:当环境条件合适(温度、pH值等)时,蛋白质折叠后的稳定三维结构完全由其氨基酸序列决定。这就是影响深远的安芬森假说(Anfinsen's dogma),它背后的深意是:尽管蛋白质折叠的过程十分复杂,其中有各种力、分子的相互作用,但所有的信息竟然都包含在了其最初的氨基酸序列之中,这个过程又是如何发生的呢?于是“蛋白质结构预测问题”,即给定蛋白质的氨基酸序列,输出其最终折叠后的三维结构,成了分子生物学中的一座圣杯。
世界各地的生物学家拿起计算机这件强大武器,设计各路算法来追逐这座圣杯。这里有一个问题:为什么不直接通过实验观测蛋白质来确定其空间结构呢?事实上,自1970年代以来,通过实验方法来确定蛋白质结构在不断进步,精度不断提高,特别是冷冻电镜技术得到应用后,结构生物学得到了长足的发展。通过实验方法确定的蛋白质结构也被认为是标准答案。不过这些实验方法非常耗时耗力,据估计,用实验方法确定一个蛋白质结构需要10万美元和长达数月的时间[2]。因此,如果能设计一个计算机算法来预测蛋白质三维结构,那将会大大地加速蛋白质结构的分析。并且计算机科学和算力的飞速发展也给生物学家们敞开了一扇新的大门,但是,用计算机算法来做预测绝非易事。
计算机预测的两种方式
使用计算机来对自然过程的结果进行预测通常有两种方式。第一种方式基于物理学,模拟计算分子运动过程,我们不妨称之为“模拟派”。这种方式在科学计算领域有广泛应用,我们每天都看的天气预报正是通过计算机来模拟大气运动实现的。类似地,看过《三体》的读者们一定都知道,三体运动不存在精确的解析解,但可以通过计算机进行数值模拟:将连续的物理过程模拟成很多离散的小步,然后在每一小步计算出可以接受的近似解,依次迭代完成最终的模拟。然而,要高分辨率地模拟物理过程需要巨大的计算量。
在蛋白质结构预测领域,Folding@Home便是一项知名的“模拟派”项目,它由斯坦福大学教授 Vijay Pande 于2000年发起,联合全世界志愿者的计算机来构成一个超大的分布式计算机,它同时也是全世界第一台 exaflops 级别(每秒进行1018次双精度浮点数计算)的超级计算机。这样强大的算力使得 Folding@Home能够对蛋白质折叠过程进行原子级别的模拟,超出先前估计可模拟的时段数千倍,其成果已经参与了200余篇科学论文的发表。
模拟法的优势在于它可以模拟出完整的、动态的分子运动过程;但是另一方面,由于需要巨大的计算量,对于比较大的蛋白质,模拟出最终的三维结构需要非常长的时间。不过,正是因为“模拟派”试图模拟蛋白质折叠完全的过程,这使得他们的工作的意义远远不止预测最终的三维结构。试想,若是计算机能将蛋白质折叠完整的动态过程如动画般展现出来,这对于科学家理解蛋白质的错误折叠和药物设计都会有极大帮助。正因如此, Folding@Home在对于蛋白质错误折叠疾病的理解和治疗上已经做出了若干贡献。
计算机预测自然过程的第二种方法是基于统计规律的预测,我们可以称之为“统计派”。“统计派”不直接对物理过程进行模拟,而是将其视作一个黑箱,通过对过往的输入和输出寻找规律,进而对新的输入进行预测。换言之,这个过程是“数据驱动”的,首先需要有过往历史数据,然后从数据中寻找规律。因为不需要模拟黑箱中完整的物理过程,“统计派”显然比“模拟派”在计算上更高效。还是以天气预报作为例子,“统计派”从过往的天气中来寻找规律和特征,运用数理统计方法对未来做出预报,这就是天气预报中的数理统计预报法。
然而对于蛋白质结构预测的问题,想要精确地找出输入输出之间的规律也十分困难,因为理论上任意两个氨基酸之间都可能发生相互作用,其中的物理化学过程十分复杂。此外,“统计派”还有一个致命的难点在于,当过往数据中缺乏与当前输入类似的输入时,预测将会变得更加困难。
AlphaFold为何如此耀眼?
“统计派”的一大巨作便是DeepMind公司开发的AlphaFold了。其实在AlphaFold横空出世之前,其他的预测算法(如 Rosetta@Home)也一直在进步,准确率不断提升,然而AlphaFold的光芒实在太耀眼,以至于前人算法的进步显得十分暗淡。
2020年,AlphaFold 2参加了蛋白质结构预测技术关键测试(CASP)比赛。这是蛋白质结构预测界的奥林匹克竞赛,来自世界各地的参赛团队会拿到未知结构的蛋白质的氨基酸序列,然后使用自己的算法预测其三维结构,最后和实验测定结果进行比较,相似度越高分数就越高。在这一年的比赛中,AlphaFold 2取得了中位数分数92.4分(满分100分;90分以上被认为预测方法可与实验方法媲美)的高分预测结果,它预测的蛋白质三维结构和最后实验观测的标准答案,仅有原子大小尺度的差异!这样出色的成绩以至于让很多人认为,这个困扰了生物学界50年的问题就这样被解决了!当然,“圣杯”不会那么容易被拿下,但生物学家迎来了一件利器。
AlphaFold是怎么做到如此准确的预测的呢?它的核心武器就是深度学习。深度学习是一种从2010年代开始蓬勃发展的机器学习技术,它通过建立多层的神经网络来模拟人脑的学习方式,具有强大的泛化能力和表达能力,能够自动地从过往数据中学习复杂的模式与特征,以此对于新输入进行精准预测。事实上,在2018年的CASP比赛中AlphaFold便已经参赛并夺得头筹,不过那时还尚未达到如此高的预测准确率。
不过,AlphaFold 2同样也面临诸多挑战。首先,深度学习通常需要庞大的数据集来进行学习,而当时的蛋白质数据库(Protein Data Bank)中只有大约17万条蛋白质数据。这听起来似乎已经不少了,但是我们横向对比一下就知道这个数据量有多么捉襟见肘:同样是在2020年训练的语言模型GPT-3使用了3000亿个token(可以理解为“词”)作为数据集,这可比17万大了许多个数量级。其次,AlphaFold 2预测出的三维结构要遵循基本的空间几何规律。从分子结构来说,三个原子所构成的三角形应该遵循两边之和大于第三边的基本规律,而这样的规律又应该如何与机器学习模型训练的过程结合起来?换言之,如何让机器在有限的数据集中习得这些知识呢?
AlphaFold预测蛋白质基本原理
图2是AlphaFold 2深度学习模型架构示意图。最左边的输入表示需要被预测结构的序列(input sequence);旁边画了一个小人,代表人类的某种蛋白质。
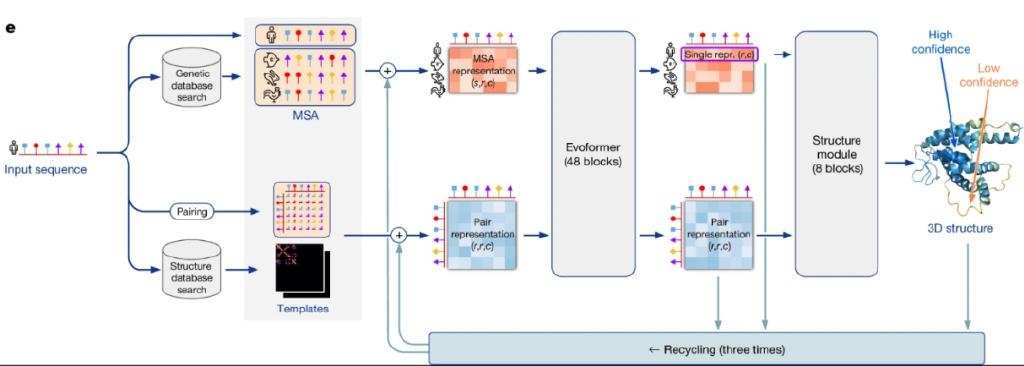
图2:AlphaFold 2深度学习模型架构图
接下来,这个输入序列被转换成两种不同的信息,传入后面的神经网络进行迭代和学习。第一个信息,上面的“MSA”是多序列对比(Mutiple Sequence Alignment)的缩写,意思是说,我们从一个现有的基因数据库中搜索出与当前输入序列最接近的一些序列进行对比。当然,搜出来的这些序列不一定是存在于人体中的,图中就举例了三个类似的序列,分别画上了鱼、兔子和鸡,代表其来自相应的生物体。搜索这些类似的氨基酸序列有什么用呢?这是因为生物学里有一些基本的遗传规律,通过观察其他存在的相似的序列,我们可以推测两个氨基酸之间是否存在关联,而这对于预测最终的三维结构是非常有用的信息。举个例子,我们可能发现某两个氨基酸(譬如一个在序列首位、一个在末尾)总是成对地出现突变(mutation),这就意味着它们俩在最后的三维结构中会有某种关联。
在基因数据库中搜出来了以后,MSA信息会被转换成一个矩阵,也就是图片中橙色的矩阵(MSA representation)。这个矩阵的维度是(s,r,c),s代表序列数,r代表氨基酸序列的长度,c代表氨基酸embedding的长度,相当于我们用一个向量来表示一个氨基酸,其长度就是c。这个MSA representation矩阵在模型的训练过程中会被反复迭代和更新。
第二个用来学习和迭代的信息是图2中的“Pair Representation”,它是一个维度为(r,r,c)的矩阵,相当于把输入序列横着和竖着排列起来(行和列),形成一个正方形的矩阵;而元素(i,j)代表氨基酸i和氨基酸j之间的空间关系,如距离、角度等,用一个长度为c的向量来表示。换言之,这里用了两个不同的矩阵,一个是MSA,代表生物演化信息;另一个是Pair,代表空间几何信息。在模型训练的过程中,这二者相辅相成、相互更新,使得模型能够充分融合、汇总两者包含的信息。
MSA和Pair Representation被传入Evoformer模块中进行更新,图3是DeepMind论文中提供的Evoformer展开图。如它的名字所暗示,Evoformer是Transformer的一个变种,即在普通的Transformer模块里加入了各种MSA与Pair representation相互融合的操作,以及一些特定的模块使得机器能够习得空间几何关系,等等。关于Transformer已经有许多介绍,这是一种十分强大的神经网络架构,特别擅长捕捉长序列的上下文关系(long-range dependency),ChatGPT名字里的T正是Transformer。
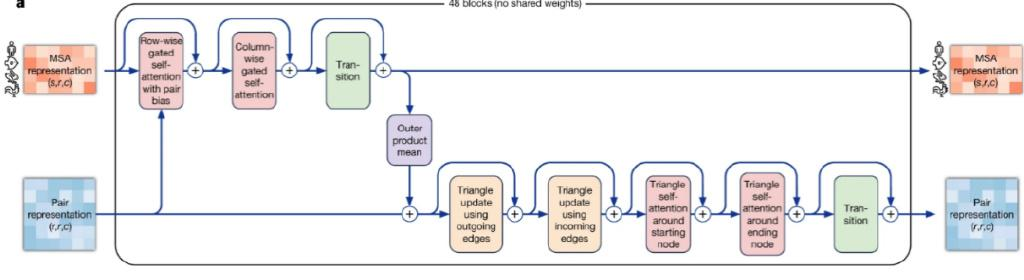
图3:Evoformer模块展开图
Transformer的核心是一种被称为自注意力(Self-Attention)的机制,它使得模型能够学习出一段序列中每一个元素和其他元素的相关程度的高低。在语言模型的应用中,它能帮助模型学习到一句话中每一个词和其他词的相关性。在蛋白质结构预测的应用中,输入的氨基酸序列恰恰也是一种序列——氨基酸按顺序排列在一起,互相之间可以发生相互作用,这使得自注意力机制十分适用于这个场合。这也充分显示了Transformer的通用性,只要是可以表示为序列的数据,不管是文字、语音、图像,还是蛋白质序列,都可以使用Transformer来捕捉上下文信息。
原始的Transformer处理的是文字这种一维的序列,而AlphaFold两种不同的输入(MSA和Pair representation)都是二维的矩阵,于是在Evoformer模块中,自注意力是以轴向注意力(Axial attention)的方式被使用,它是自注意力的一个变种,适用于对超过一维的数据进行上下文捕捉,即每次按某一个轴来进行那个维度上的自注意力计算。对于矩阵(二维)而言,是按行(row-wise)和列(column-wise)进行自注意力计算。
Evoformer模块里还包括了若干用来满足三角不等式(即三角形的两边之和大于第三边)的模块,譬如Triangle multiplicative update(黄色框)和Triangle self-attention(粉红色框),两者皆可用来帮助模型学习到三角不等关系。前者需要的计算量更小,但AlphaFold 2团队发现,两者一起用预测准确率更好。这样的Evoformer模块一共有48个,使得模型可以一层一层地学到深层次的特征模式,在此过程中,不断地更新MSA和pair representation,使得它们能够表征更加丰富、准确的特征信息。
我们前面提到,Alphafold 2的挑战之一就是数据集比较小。研究人员的解决方案是,除了将MSA等演化相关的先验的人工知识嵌入到神经网络之中以外,还使用了一些扩充数据集的技术,如Self-Distillation——他们使用了一个未标注的、具有约35万条氨基酸序列的新数据集Uniclust30,并使用AlphaFold 2对其进行预测,然后通过将其中高置信度的预测结果加入到原始PDB数据集中,形成了一个扩充后的新数据集,然后在这个新的数据集上对模型进行重新训练,这样做也提升了最后的预测准确率。
总体而言,在训练集不是很大的情况下,AlphaFold 2的一大设计挑战就是将人工的知识和模型学习的过程有机地融合在一起。AlphaFold 2也很好地应对了这个挑战,最后模型取得了出色的预测效果,掀起了一场生物信息学的革命。
更通用的AlphaFold 3
AlphaFold的脚步并没有止步于AlphaFold 2,就在2024年5月,DeepMind推出AlphaFold 3,再次引起业界轰动。AlphaFold 3不仅仅能够预测蛋白质的三维结构,也能预测更广泛的生物分子复合物的结构(包括蛋白质、核酸、配体等),以及生物分子之间的相互作用。有趣的是,尽管AlphaFold 3对于预测精度和广度都有提升,它自身的模型架构相比AlphaFold 2却更加简化而且通用了。AlphaFold 2虽然使用了Transformer这种通用的模型,但同时它也加入了许多工程化的细节去提升最后预测的结果。而在AlphaFold 3中,工程化的细节得到了简化,取而代之的是更强大、更通用的模块。譬如AlphaFold 3大大降低了对于MSA的使用和依赖,而主要使用Pair representation进行学习。AlphaFold 2中的核心模块——Evoformer的数量在AlphaFold 3中被减到了4个;同时AlphaFold 3也增加了一个新的模块Pairformer,而这个模块只对Pair representation进行更新,其数量是48个。这样的设计是为何呢?如我们前文所述,MSA中保存的是从基因数据库中搜出来的氨基酸序列,其包含了蛋白质演化信息,而Pair representation包含的是空间几何信息。毫无疑问,后者是一种更加通用的表达方式,它不仅限于蛋白质这一种分子,也适用于配体等其他分子。因此,降低对MSA的依赖而加强对Pair representation的学习与更新能使得模块更加通用化、能处理更多不同类型的输入,这也正是AlphaFold 3的目标。
AlphaFold 3另一通用化的设计在其结构模块(structure module),即从Pair representation生成最终的三维结构图。上一代的AlphaFold 2把蛋白质最终的三维结构视为一系列由氨基酸残基构成的三角形在空间中的旋转和平移构成的主干框架(backbone frames)以及侧链的扭转(side-chain torsion),通过让模型计算出这些三角形的旋转角度、平移大小以及侧链的扭转角度来得出最终的蛋白质三维图像,这其中的技术细节十分复杂。而AlphaFold 3删繁就简,模型架构中不再手动地编入三角形、旋转、平移等概念,而是直接采用了一个标准的扩散模块(diffusion module),这个扩散模块将直接预测每一个原子在三维结构中的坐标,这也使得AlphaFold 3不再仅仅局限于蛋白质这一种分子的结构预测了。令人惊奇的是,尽管AlphaFold 3在模型架构中去掉了许多“蛋白质特定”的部分,其在“蛋白质结构预测”这一问题上的预测结果竟然也超越了AlphaFold 2。这似乎意味着,当模型走向通用化的时候,它以自己的方式从更加多样化的输入和任务中学习到了一些更本质的物理、化学规律,其海量的矩阵中,似乎包含了对我们所生活的世界更精准的认知。而这些更广博的知识也使得AlphaFold 3在专项任务上的表现更加出色。
最后总结一下,AlphaFold系列通过精准预测出最终的蛋白质结构,成为科研工作者的有力帮手;也再次证明了深度学习从数据中学习复杂模式的能力,借此AI变得更加智能与多任务化。尽管如此,人类对于蛋白质折叠完整的动态过程和机理仍然需要更多的理解,但AlphaFold已经成为了AI推动科学发展的一座里程碑,而愈发强大的AI对于科学研究的助力还将继续。

特 别 提 示
1. 进入『返朴』微信公众号底部菜单“精品专栏“,可查阅不同主题系列科普文章。
2. 『返朴』提供按月检索文章功能。关注公众号,回复四位数组成的年份+月份,如“1903”,可获取2019年3月的文章索引,以此类推。
版权说明:欢迎个人转发,任何形式的媒体或机构未经授权,不得转载和摘编。转载授权请在「返朴」微信公众号内联系后台。



 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国