

导读
近日,哈佛大学的Eric N. Jacobsen教授课题组在Nature上报道了小分子(646 Da)氢键供体(HBD)催化剂通过重现酶采用的几何预组织原理来加速对映选择性Michaelis-Arbuzov反应的SN2步骤。机理和计算研究表明,HBD降低了氯亲核试剂的反应性,但通过将磷鎓阳离子和氯阴离子重组为准备进入SN2过渡态的几何结构,加速了决定速率的脱烷基化步骤。这种新的对映选择性Arbuzov反应可高对映选择性地获得一系列H-膦酸酯,其可进行多种衍生化。该工作首次展示了对磷鎓脱烷基化步骤的催化对映控制,为P-立体异构化合物的合成建立了一个新平台。文章链接DOI:10.1038/s41586-024-07811-4。

图1. 将酶策略应用于离子SN2机制的催化
(图片来源:Nature)
正文
化学加——科学家创业合伙人,欢迎下载化学加APP关注。
双分子亲核取代(SN2)机制在有机化学领域的历史发展和教学中占据着核心地位。带电物质之间的双分子SN2途径在极性非质子溶剂中通常比在质子溶剂中进行得更快(图1A)。而涉及离子SN2机制的反应的选择性催化必须克服这样一个事实:任何与离子对的明确静电相互作用都可能使反应物相对于过渡态稳定,并导致速率衰减而不是加速。
在促进SN2机制的少数酶类中,大多数采用一般酸碱催化来激活不带电的亲核试剂或亲电试剂。亲核卤化酶5'-氟-5'-脱氧腺苷合酶(FDAS)的另一种催化模式已有报道,其促进阳离子S-腺苷甲硫氨酸(SAM)上的氟化物(或氯化物)置换 (图1B)。固态X-射线结构表征(图1B)以及立体化学、理论和动力学研究已确定该酶通过卤化物结合活性位点实现速率加速~106 (i)将卤化物精确定位在与离去基团的共线关系中和(ii)提供“卤化物空穴”以抵消亲核试剂从水中脱溶的能量损失。在这种酶促机制中,酶活性位点被预先组织起来,以稳定几何上类似于过渡态的“近进攻”基态构象。作者设想了一种仿生方法来设计用于离子SN2机制的小分子催化剂,并以FDAS结构-机制关系为指导。作者假设,如果催化剂能够(i)精确地将离子对预组织成适合SN2 亲核取代的几何构型,并且(ii)为亲核试剂提供直接溶剂化壳(图1C),则成功的催化是可行的。
作者选择了Michaelis-Arbuzov反应来探索,Arbuzov反应中的关键步骤是磷中间体的 SN2脱烷基化,这一基本步骤也是有机磷化合物合成中其他多种重要反应的基础。设想的转化始于具有两个相同烷氧基取代基的非手性P(III)物种,在形成四面体膦阳离子后,它们将呈现用于SN2脱烷基化的对映位点(图1D)。如果SN2步骤是周转限制,那么实现高选择性还需要催化剂加速SN2步骤而不是外消旋未催化的反应。本文中,精确设计的手性 HBD加速了高对映选择性Michaelis-Arbuzov反应中膦卤化物离子对中间体的 SN2脱烷基化(图1C)。所得的H-膦酸酯产物是制备立体异构体-磷(V)化合物的多功能中间体。
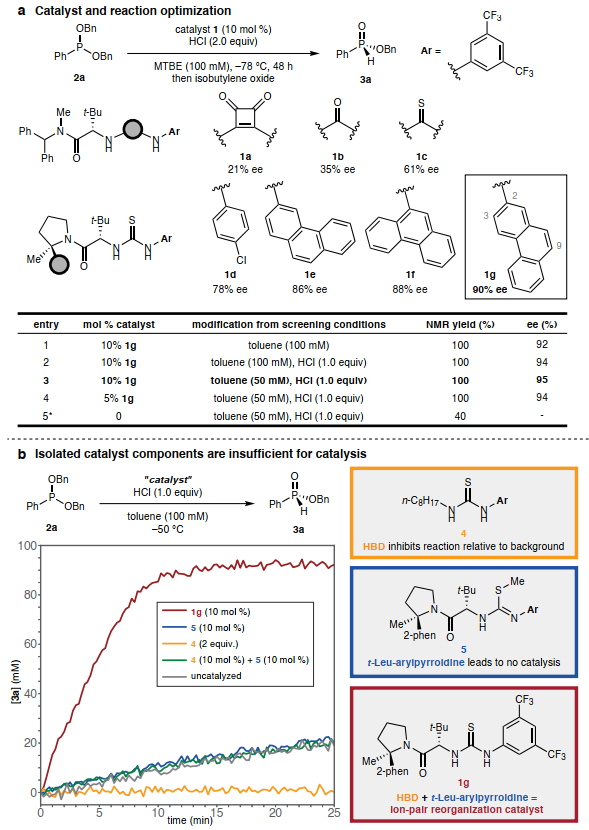
图2. 反应的发展及优化
(图片来源:Nature)
对Arbuzov反应类型与手性HBD催化剂的实证研究表明,二苄基苯基亚膦酸酯 2a与HCl的脱烷基化具有良好的对映选择性(图2A)。最终,带有2-菲基取代基的硫脲1g以90% ee和定量产率促进了脱烷基化。将反应溶剂改为甲苯(图2B),使用一当量的HCl,并将反应混合物稀释至50 mM,以95% ee得到3a,可以显著提高对映选择性。
催化剂1g使2a的反应加速了约30倍。催化剂1g的简单硫脲类似物在抑制离子对坍缩方面模仿了质子溶剂。通过硫脲的S-甲基化合成了5,从而消除了催化剂1g的双HBD特性,与背景反应相比,化合物5没有引起速率加速(图2B,蓝色)。这些观察结表明两个域的存在以及它们在1g中精确的相对空间取向对于催化是必要的。
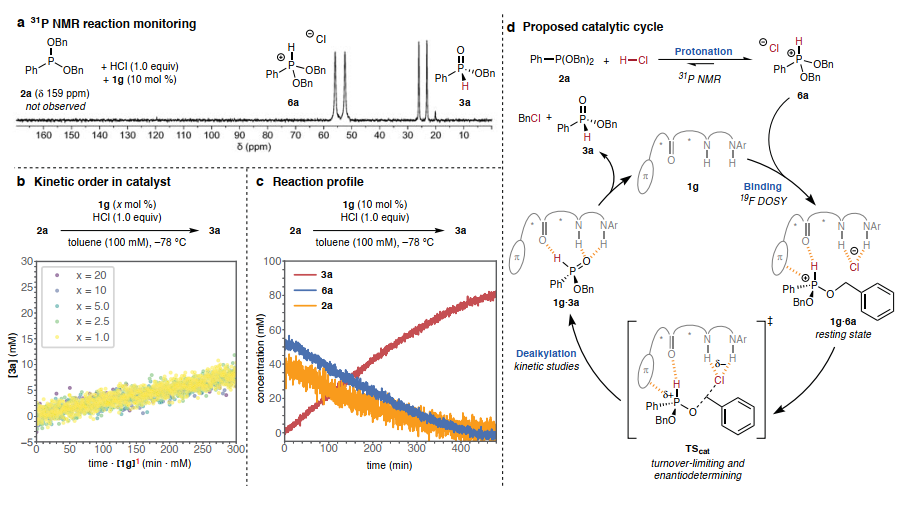
图3. 机理研究
(图片来源:Nature)
作者进行了系统的机理研究,通过31P NMR监测2a与HCl 在硫脲1g催化下的反应,获得了催化体系中存在质子膦离子的证据(图3A)。观察结果表明,氯化磷6a是催化条件下底物的静止状态。采用扩散有序核磁共振光谱法(DOSY)测量催化剂1g在催化反应条件下的扩散常数,以此作为估算其分子量的方法,研究了催化剂1g的静止态形式(图3B)。反应完成后,测量的分子量介于单体1g和1:1 1g·3a 复合物之间,与游离催化剂和产物结合催化剂之间的平衡一致,与强催化剂-产物结合不一致。这些观察结果表明,催化剂1g与6a以1:1的复合物形式静止,并且限速脱烷基化反应从该复合物进行。
在确定了静止态复合物的分子组成后,作者确定了[1g]T和[6a]的动力学依赖性,以阐明速率决定过渡态复合物的化学计量学(图3B)。结果表明反应速率表现出对催化剂[1g]T的一级依赖性。此外,磷物种6a的消耗和产物3a的形成在反应的前~80%中都遵循零级动力学速率行为(图3C),与从1:1 1g·6a 静止态复合物进行的周转限制和对映决定脱烷基化过渡态一致。图3D描绘了与所有可用机制数据一致的催化循环。该循环的特点是:(i)质子化平衡有利于氯化磷6a,(ii) 6a与单体催化剂1g结合形成静止状态,(iii)周转限制和对映决定脱烷基化形成产物3a和苄基氯,它们从催化剂上分离,从而完成催化循环。
催化剂1g的尺寸相对较小,使得SN2步骤可以通过DFT进行明确建模(图4A)。在没有催化剂的情况下,发现氯化磷6a以紧密离子对的形式静止,具有笼状结构,其中氯阴离子与阳离子发生多种稳定相互作用。必须牺牲所有稳定的 H 键相互作用和基态中存在的相当一部分库仑引力,其方式与卤化酶反应中从水介质中脱溶大致类似,以实现 SN2机制所要求的线性几何形状。在考虑这种几何重组时,协同脱烷基化途径可以在概念上分为离子对重组阶段和随后的离子对崩溃阶段。这两个阶段可以用位于计算的本征反应坐标上的非稳态6a’来划分,其中氯离子位于SN2轨迹上,但C-O键的形成尚未开始。通过此分析,>75%的整体电子活化能垒来自第一阶段中氯离子的重组,而对应于共价键断裂和形成事件的离子对坍缩阶段对整体能垒的贡献小于 25%(约 4 kcal/mol)。
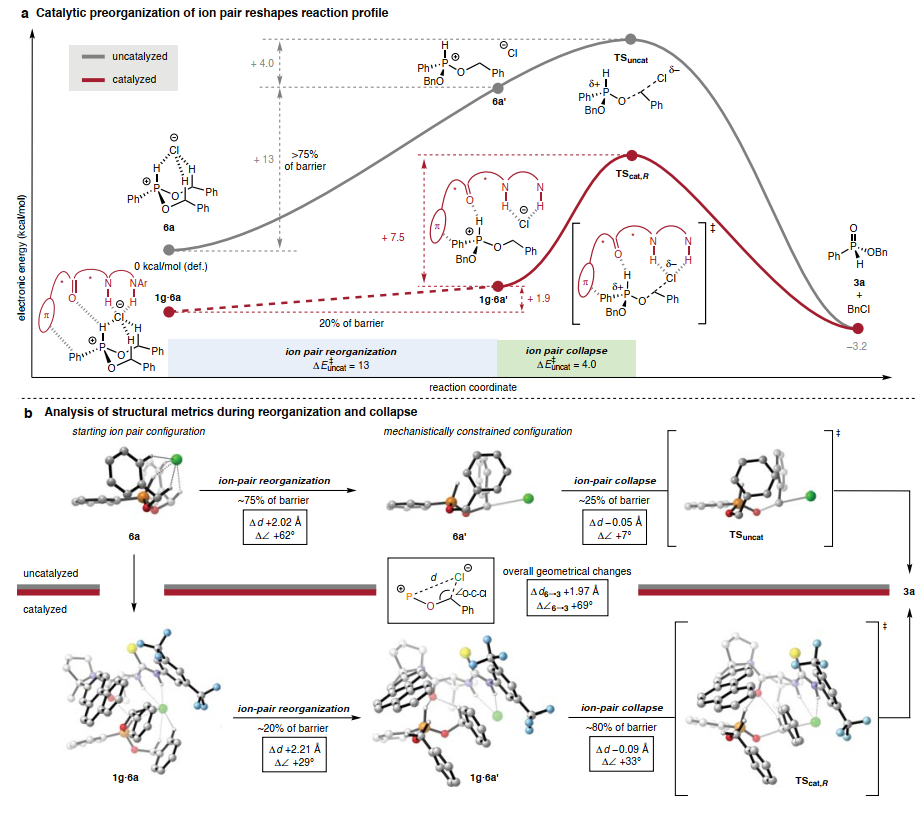
图4. 催化速率加速的起源
(图片来源:Nature)
在催化途径的计算模型中,发现亲核取代以两个离散步骤进行(图4A)。氯化磷静息态复合物1g·6a’的几何特征与6a’非常相似,即氯化物通过氢键定位在接近最佳的过渡态前几何结构中(图4B),准备进入TScat,R。因此,催化剂1g可以被视为参与了吸引性非共价相互作用网络,从而获得了相对稳定的基态复合物1g·6a’,该复合物为脱烷基化反应做好了准备。催化剂结合提高了离子对崩塌的障碍相对于非催化途径,这与预期的H 键对氯亲核性的减弱作用一致。然而,离子对崩塌的抑制被催化剂减轻了氯化磷离子对所需的几何预组织的能量成本所抵消,导致相对于背景反应的整体加速。
系统的构象搜索导致鉴定出低能非对映体结构,从而得到3a的主要(R)和次要(S) 对映体,其相对能量与实验观察到的对映选择性高度一致。导致次要 (S)和主要(R)产物对映体的非对映体过渡态结构显示出几乎相同的催化剂几何形状,但与催化剂活性位点内磷离子的120度旋转有关。虽然TScat,S和TScat,R都具有几个共同的非共价吸引相互作用,但TScat,S 在苄基C-H和酰胺氧之间具有一个额外的H键相互作用,而TScat,R 结合了两个苄基C-H–π相互作用。因此,失去一个苄基C-H氢键相互作用和获得两个苄基C-H–π 相互作用的净能量效益被认为在决定对映诱导的意义和大小方面起着关键作用。
最后,考察了底物范围。多种二苄基亚膦酸酯被证明可用作底物,提供空气和水分稳定的手性H-亚膦酸酯产品,可通过硅胶柱色谱法纯化(图5A)。各种对位取代的芳基亚膦酸酯经过脱烷基化,产率和对映选择性良好(3a-3g),对于具有强吸电子取代基的底物(3h-3j),对映选择性较低。间位取代的苯基亚膦酸酯经过脱烷基化,对映选择性与对位取代的区域异构体(3k-3m)相当。邻氟取代将对映选择性从90% (3f)降低到73% ee (3n),而邻苯基取代则降低了对映选择性(3o)。然而,带有邻位稠合多芳族取代基的底物经历了高对映选择性的脱烷基化(3p和3q)。该方法还适用于各种杂芳族和多芳族取代基(3r-3t和3v)。具有非芳基取代基的膦酸酯,可进行脱烷基化,具有中等水平的对映选择性,而烷基取代基,如环丙基和甲基,则分别具有54%和30% ee的低水平对映选择性。2a的对映选择性脱烷基化在克级规模上成功进行,仅使用3 mol % 1g,具有高对映选择性(93% ee)、产率(98%)和高效催化剂回收率(95%)。作者探索了(R)-苄基苯基亚膦酸酯3a作为正交双功能化的P-立体结构砌块的反应性。发现它适用于一系列P-H部分的立体特异性合成工艺,然后对P-OBn部分进行二次衍生化(图5B)。基于已建立的膦酸酯的磷化Mannich反应性,用苯酚锂去质子化使3a能够参与Pudovik加成 Eschenmoser盐,得到α-氨基膦酸酯7a。随后可以在甲基锂存在下取代7a的苄氧基以得到8a,从而保留磷处的对映体富集。用亲核试剂如苄胺和酪氨酸来获得具有高产率和对映特异性的膦酸酯和膦酰胺9a/b。为了研究磷酰基介导的反应性,将苯酚锂条件调整为先前报道的H-膦酸酯和苯醌之间的1,6-偶联,获得了具有优异立体特异性的O-磷酰基对苯二酚衍生物10a。该结果表明,在没有手性控制元素的情况下,3a衍生的磷酰基具有足够的构型稳定性来参与立体特异性反应。最后,对3a进行硫化-甲基化序列,得到具有优异对映特异性的硫代膦酸酯11a。
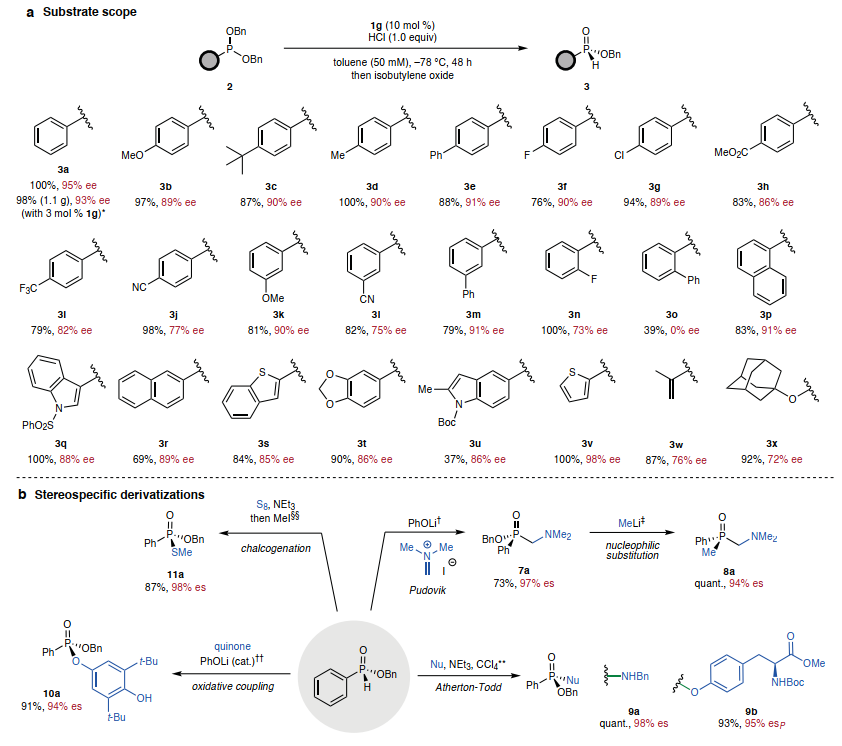
图5. 底物范围和合成转化
(图片来源:Nature)
总结
在有利的过渡态前几何结构中对反应物进行精确预组织,这是酶催化的基本机制原理,Eric N. Jacobsen教授现已在HBD催化剂的背景下得到开发和表征,用于对映选择性Michaelis-Arbuzov反应,可产生有价值的P-立体异构产物和砌块。预计这种几何预组织原理在小分子系统中将越来越重要。
文献详情:
Catalysis of an SN2 pathway by geometric preorganization.
Gabriel J. Lovinger, Marcus H. Sak & Eric N. Jacobsen*.
Nature, 2024
https://doi.org/10.1038/s41586-024-07811-4
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国