

导读
近日,美国加利福尼亚大学伯克利分校Richmond Sarpong课题组报道了一种多聚假吲哚酚(poly-pseudoindoxyl)天然产物Baphicacanthcusine A的首次全合成。该合成利用吲哚氧化重排为假吲哚酚(pseudoindoxyls),以非对映选择性的方式引入相邻的假吲哚酚杂环。关键步骤包括酸介导的环化/吲哚转位(transposition)、两次非对映选择性氧化缩环(ring contractions)和一次位点选择性C-H氧化反应。文章链接DOI:10.1002/anie.202409139
(图片来源:Angew. Chem. Int. Ed.)
正文
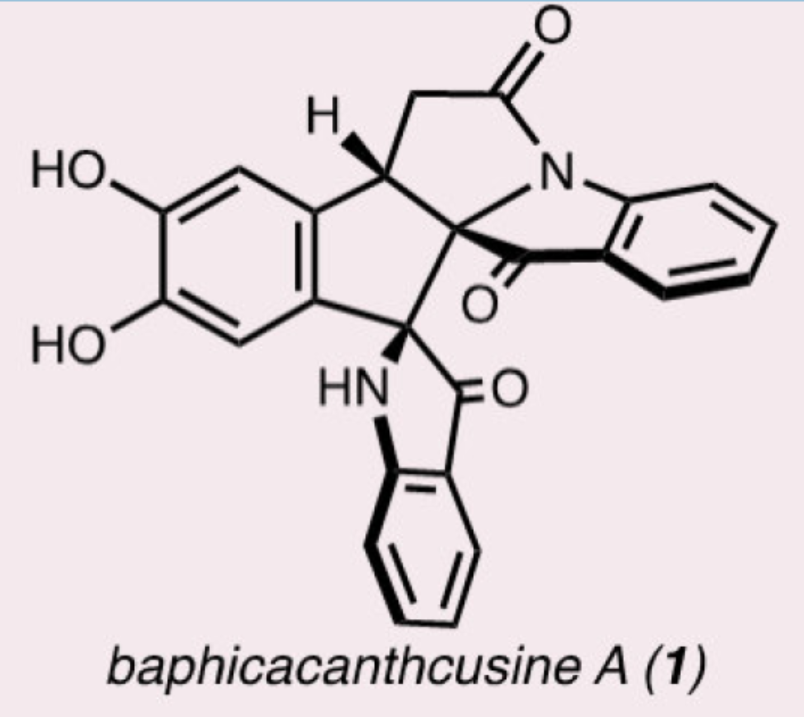
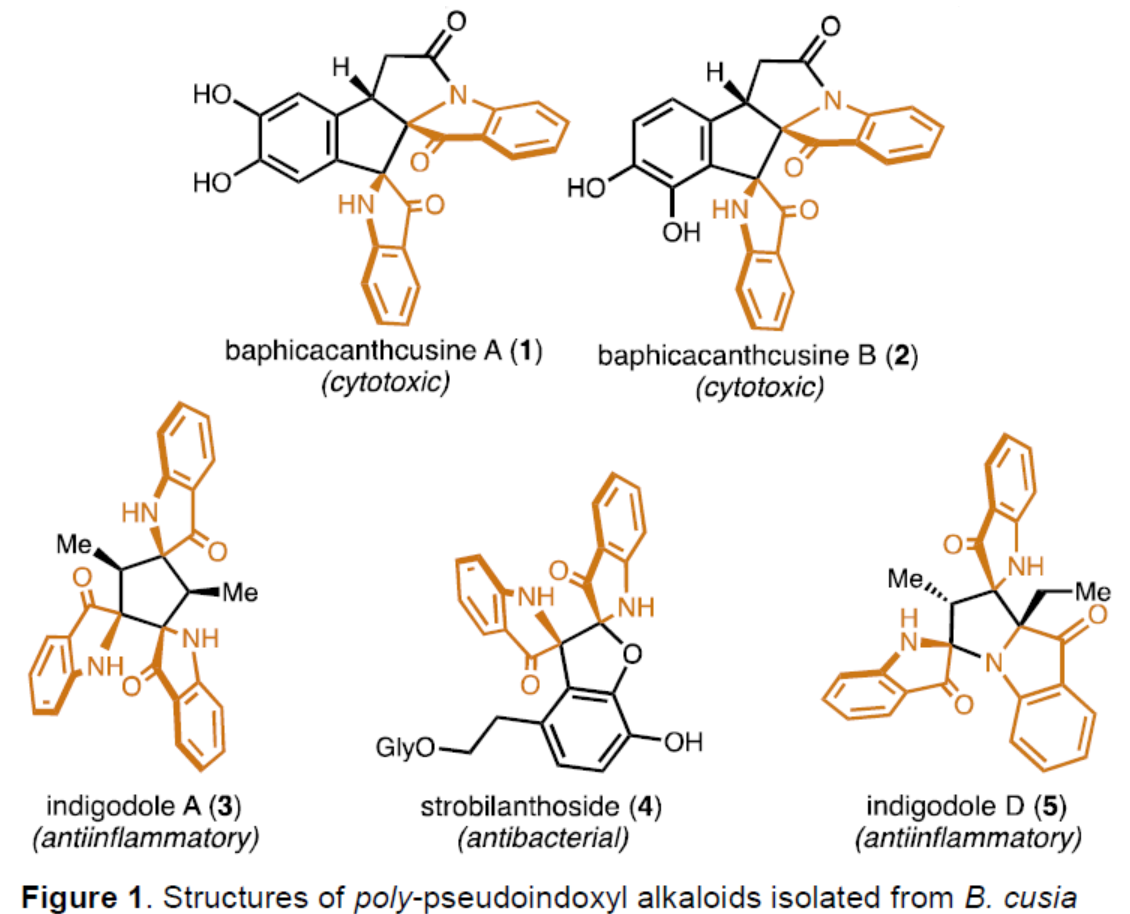
几个世纪以来,开花植物Baphicachanus cusia(Nees)Kuntze或Strobilanthes cusia(Nies)Bremek一直作为东亚传统药物的成分。B. Cusia的叶子、茎和根可用于治疗多种疾病,如牛皮癣、白血病和普通感冒,这促使化学家们寻找负责这一系列药理活性的特定化合物。最近,化学家们已从B. Cusia中分离出了多种多聚假吲哚酚生物碱(1-5,Figure 1),其特征是中心五元环带有两个或三个假吲哚酚杂环。尽管存在这种共同的骨架,但多聚假吲哚酚生物碱表现出显著的结构和药理学多样性,为合成和生物探索提供了充足的机会。迄今为止,尚未实现多聚假吲哚酚生物碱家族的成员的合成。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
(图片来源:Angew. Chem. Int. Ed.)
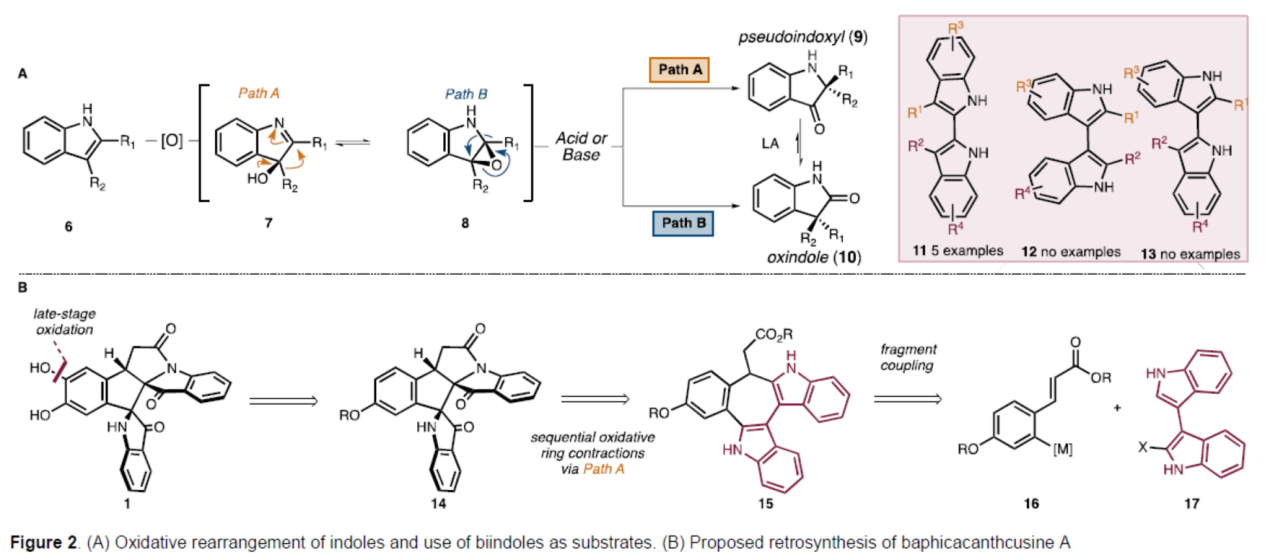
目前,化学家们已经开发了几种合成假吲哚酚骨架的策略。其中,吲哚氧化重排为假吲哚酚是最常见的方法。通常,这种转化涉及吲哚(6)氧化为羟基吲哚啉(hydroxyindolenine,7),然后通过酸或碱介导的立体专一性重排,以构建假吲哚酚(9)(Figure 2, Path A)。尽管其效率很高,但这种形成假吲哚酚的策略在氧化过程中的非对映选择性很差,在重排步骤中的选择性也很差(Figure 2A, Path B)。值得注意的是,虽然氧化重排方法已成功应用于许多全合成,但联吲哚底物的使用极为罕见。迄今为止,该策略仅用于2,2'-联吲哚(11),没有使用3,3'-或2,3'-联二吲哚(分别为12和13)的报道。此外,只有两个非对称底物的例子(即R1 ≠ R2或R3 ≠ R4)。基于上述的讨论,Sarpong课题组选择了Baphicachancusine A(1)作为目标分子,这是在2020年分离出的一种细胞毒性生物碱。Baphicachancusine A(1)含有B. cusia次生代谢产物中特有的中心五元环和相邻假吲哚酚环。此外,1带有一个与假吲哚酚稠合的内酰胺环,假吲哚酚与三个连续的手性中心和氧化敏感的邻苯二酚单元一起构成了巨大的合成挑战。在关键合成设计中,作者试图利用吲哚的氧化重排以区域、化学和立体选择性的方式引入相邻的假吲哚酚骨架。
首先,作者进行了逆合成的分析(Figure 2B)。化合物1可逆推至砌块14,后者通过C-H羟基化,可引入邻苯二酚单元。而砌块14则由砌块15通过两次迭代氧化重排合成,并可实现中心五元环的构建。砌块15可由砌块16与砌块17通过偶联合成。
(图片来源:Angew. Chem. Int. Ed.)
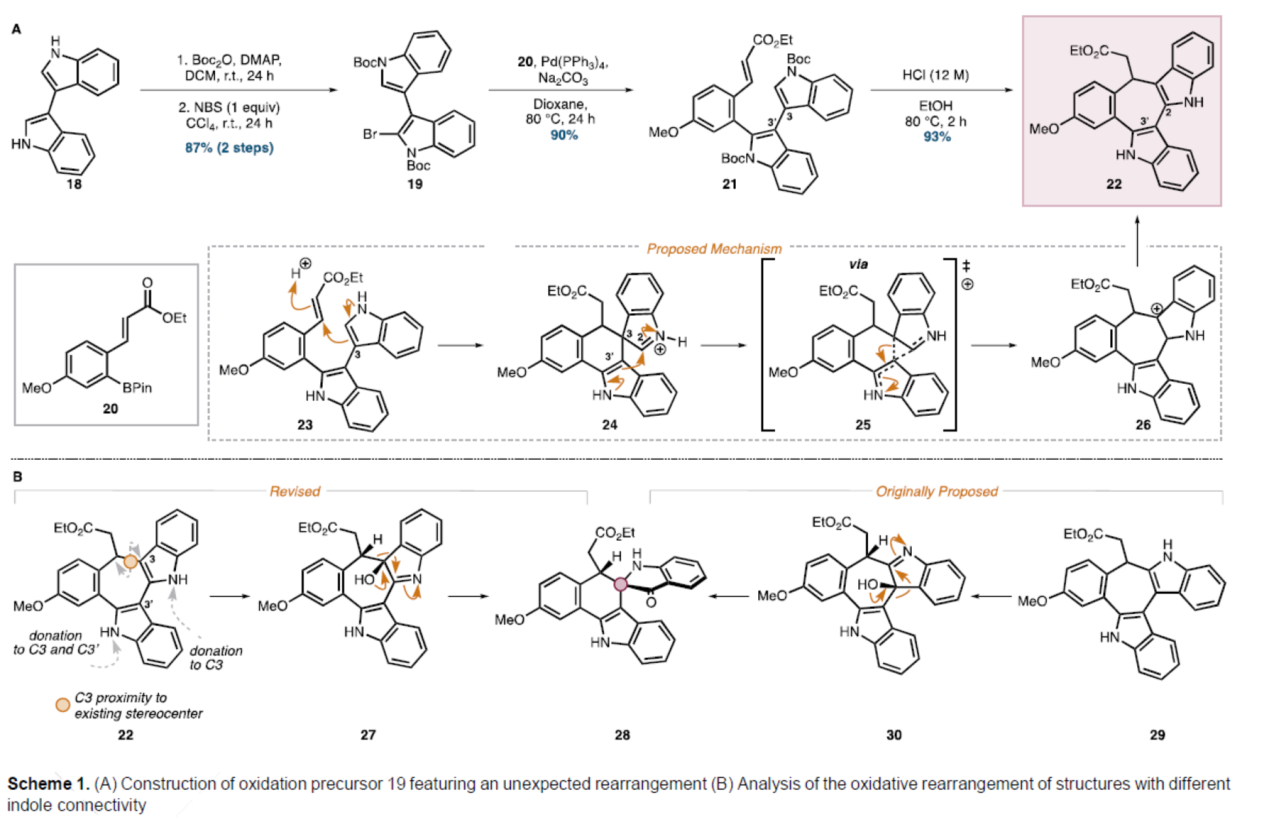
砌块22的合成(Scheme 1)。以3,3'-联吲哚18为底物,在DMAP/Boc2O/DCM条件下进行胺基的保护,并在NBS/CCl4条件下进行溴化反应,可以两步87%的总收率得到中间体19。中间体19与频哪醇硼酸酯20在Pd(PPh3)4/Na2CO3/Dioxane条件下进行Suzuki偶联反应,可以90%的收率得到环化前体21,为关键七元环中间体22的形成奠定基础。中间体21在HCl/EtOH条件下进行Boc脱保护、环化和吲哚转位反应,可以93%的收率得到七元环中间体22。值得注意的是,这种重排可能是通过北吲哚的亲核C3碳的共轭加成反应产生螺环亚胺离子24而发生的。在该阶段(通过过渡态25),C3-C3'键的迁移将产生阳离子26,阳离子26可以芳构化以生成重排的骨架(22)。通过进一步分析发现,与最初提出的底物29相比,从六环中间体22中更容易获得所需的环收缩产物28(Scheme 1B)。这种产物结果的汇聚性是因为螺稠合双环的形成仅取决于吲哚氧化的立体选择性和迁移的区域选择性,而不取决于起始稠合吲哚的连接性(27→28和30→28)。
(图片来源:Angew. Chem. Int. Ed.)
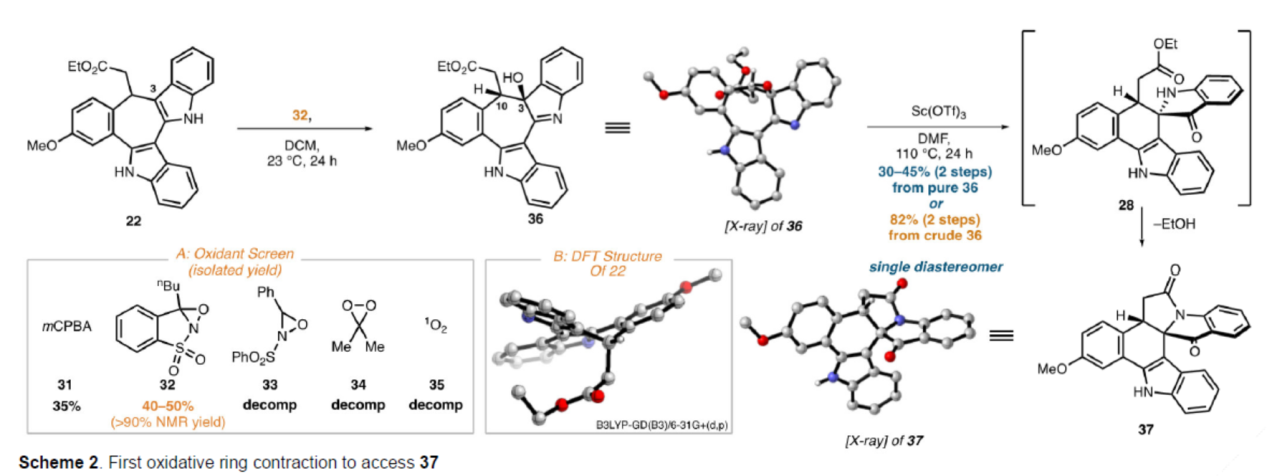
砌块37的合成(Scheme 2)。通过对中间体22氧化条件的大量筛选后发现,氧杂氮丙啶衍生物32/DCM作为最佳氧化条件,可以99%的转化率得到羟基吲哚啉36,为单一的非对映异构体,分离产率<50%。作者认为,36的低分离产率可能是由于其在硅胶上的不稳定性造成的,因此选择使用36直接进行下一步反应,而不进行纯化。中间体36在Sc(OTf)3/DMF条件下进行重排以及随后的内酰胺形成,可以两步82%的总收率得到假吲哚酚37。
(图片来源:Angew. Chem. Int. Ed.)
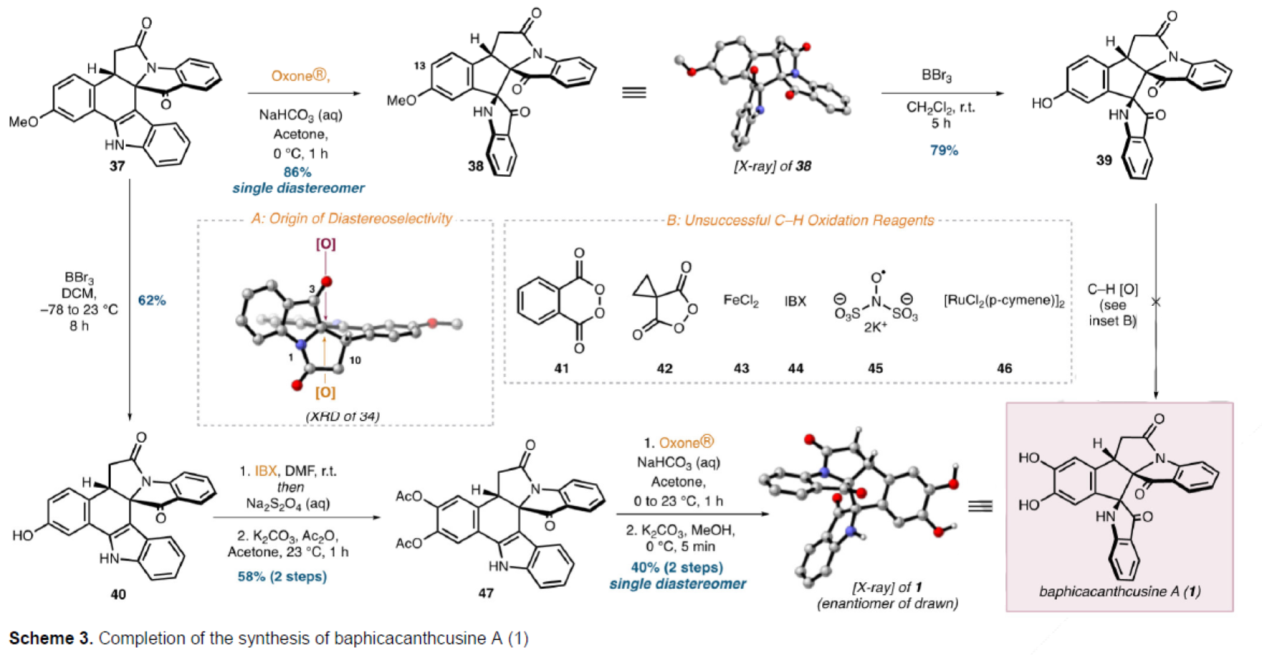
Baphicacanthcusine A的全合成(Scheme 3)。中间体37在Oxone®/NaHCO3/Acetone条件下进行氧化重排,可以86%的收率得到假吲哚酚中间体38,是单一的非对映异构体。中间体38在BBr3/CH2Cl2条件下进行去甲基化反应,可以79%的收率得到苯酚中间体39。然而,中间体39在大量C-H氧化的条件下,均未能获得目标产物1。因此,作者重新设计了另一种合成路线。中间体37在BBr3/CH2Cl2条件下进行去甲基化反应,可以62%的收率得到苯酚中间体40。中间体40在IBX(2-碘氧基苯甲酸)/DMF条件下可氧化为相应的邻醌,并使用Na2S2O4进行原位还原生成所需的邻苯二酚,随后在K2CO3/Ac2O条件下进行双乙酰化反应,可以两步58%的总收率得到中间体47。中间体47在Oxone®/NaHCO3/Acetone条件下进行氧化重排,并在K2CO3/MeOH条件下进行醇解,可以两步40%的总收率得到Baphicacanthcusine A(1),是单一的非对映异构体。
(图片来源:Angew. Chem. Int. Ed.)
总结
Richmond Sarpong课题组报道了一种简洁的路线来制备复杂的双假吲哚酚生物碱Baphicachancusine A(1)。其中,以已知的联吲哚18为初始底物,涉及11步反应。该合成是通过快速构建一个关键的七元环中间体实现的,然后通过连续的氧化和缩环重排,具有显著的底物控制的非对映体和化学选择性。对1的合成利用了联吲哚和烯酸酯砌块的汇聚性偶联,这先于意想不到的酸介导的北部吲哚基团的转位。值得注意的是,这种转位通过将初始氧化位点放置在现有手性中心附近,为高度选择性的氧化重排奠定了基础。总的来说,这项工作为引入相邻的假吲哚酚提供了一种策略,这可能会为双假吲哚酚家族天然产物的其他成员的合成提供参考。
文献详情:
Total Synthesis of (±)-Baphicacanthcusine A Enabled by Sequential Ring Contractions.
Paul P. Sinclair, Richmond Sarpong*.
Angew. Chem. Int. Ed. 2024
https://doi.org/10.1002/anie.202409139



 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国