

导读
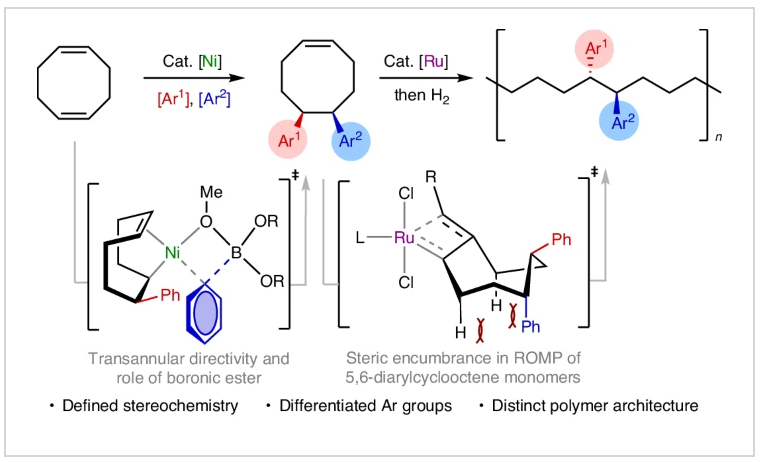
最近,美国斯克里普斯研究所(The Scripps Research Institute)Keary M. Engle课题组,美国乔治亚理工学院(Georgia Institute of Technology)Will R. Gutekunst课题组以及美国匹兹堡大学(University of Pittsburgh)刘鹏课题组报道了镍催化1,5-环辛二烯的双芳基化反应,实现了5,6-二芳基环辛烯单体的模块化合成。此转化具有良好的区域选择性和对映选择性,实现了一系列可开环复分解聚合的二芳基环辛烯单体的合成。此单体通过聚合所得到的1,2-二芳基取代的聚合物具有未曾有文献报道的头-头相连序列,且具有独特的可调节特性。机理研究表明硼酸酯在促进迁移插入中起到了对映诱导的重要作用。相关成果发表在Nat. Synth.上,文章链接DOI:10.1038/s44160-024-00618-1。
(图片来源:Nat. Synth.)
正文
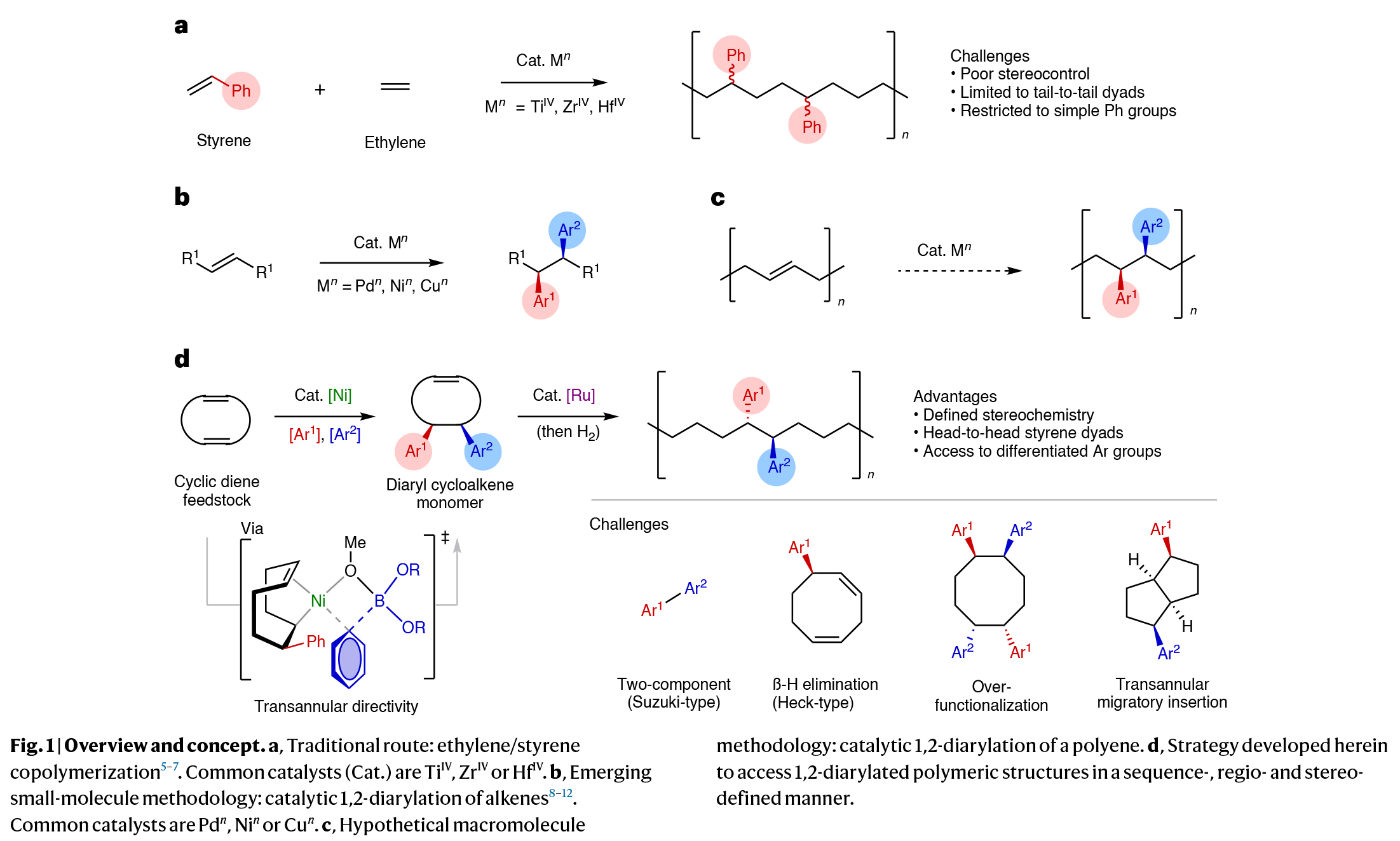
序列可控且具有特定立体构型的聚合物具有调控材料性质的应用潜力,然而它们的合成仍然具有很大的挑战性。由于聚苯乙烯和乙烯/苯乙烯共聚具有高熔点、高结晶度和低介电常数等性质,因此是重要的大分子骨架。然而,这些聚合物的合成受到现有合成技术的限制。由于芳基取代基主要局限于非取代苯环,并且相对立体化学并不明确,因此聚合反应的区域化学仅局限于尾-尾相连。因此发展可模块化、高选择性和序列可控的方式来构建未知结构的新方法备受化学家们关注。其可以有效拓宽新单体的合成思路,并可以提高聚合物结构的复杂性。最近,美国斯克里普斯研究所Keary M. Engle,美国乔治亚理工学院Will R. Gutekunst和美国匹兹堡大学刘鹏课题组联合报道了镍催化1,5-环辛二烯的双碳官能团化反应,实现了5,6-二芳基环辛烯单体的合成。该反应以模块化、区域选择性和对映选择性的方式进行,合成的单体可通过钌催化的开环复分解进行聚合,且产生的聚合物具有头-头相连的序列(Fig. 1)。欢迎下载化学加APP到手机桌面,合成化学产业资源聚合服务平台。
(图片来源:Nat. Synth.)
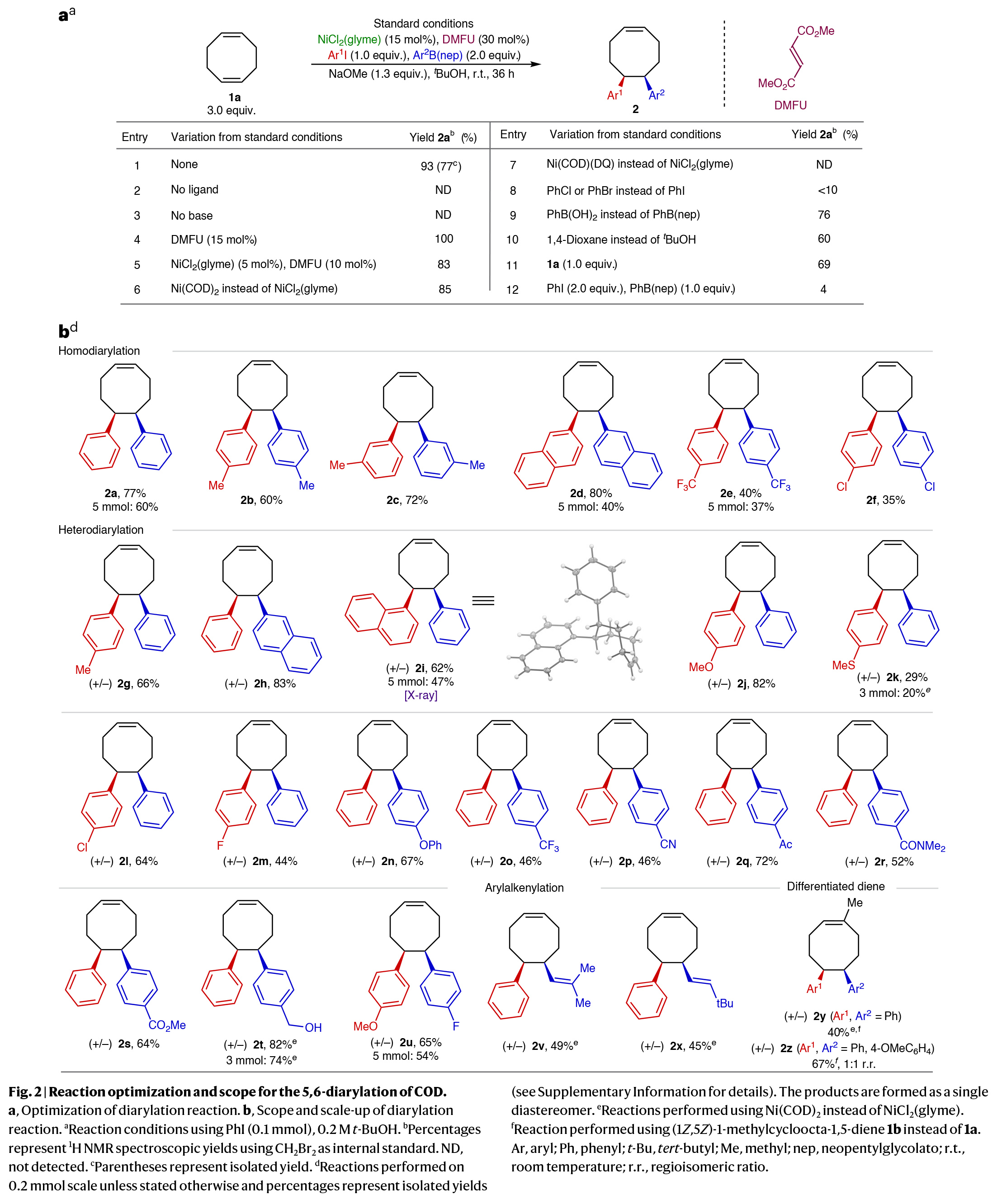
首先,作者以环辛二烯(COD) 1a作为模板底物对反应进行了条件优化(Fig. 2a)。实验结果表明,当使用1a(3.0 equiv),NiCl2(glyme)(15 mol%),DMFU (30 mol%), PhI(1.0 equiv),PhB(nep)(2.0 equiv),NaOMe(1.3 equiv),在叔丁醇中,室温反应36小时,可以以93%的核磁产率,77%的分离产率得到双碳官能团化产物2a(entry 1)。在得到了最优反应条件后,作者进行了底物范围考察(Fig. 2b)。实验结果表明,一系列不同取代的芳基碘和芳基硼酸酯均可顺利参与反应,以29-82%的产率得到相应的双芳基化产物2a-2t。其中包括一系列合成有用的官能团如氰基(2p)、酮(2q)、叔酰胺(2r)和甲酯(2s)等均可兼容。值得注意的是,此转化可以放大至3-5 mmol规模而产率基本不受影响,证明了此转化的实用性。此外,当使用烯基硼酸酯作亲核试剂时,可以分别以49%和45%的产率得到1,2-芳基烯基化产物2v和2x。最后,当向其中一个环烯烃中引入额外的取代基时,双芳基化过程会选择性地发生在位阻较小的C=C键上(2y),而此底物的杂二芳基化过程则得到了1:1的位置异构体(2z)。
(图片来源:Nat. Synth.)
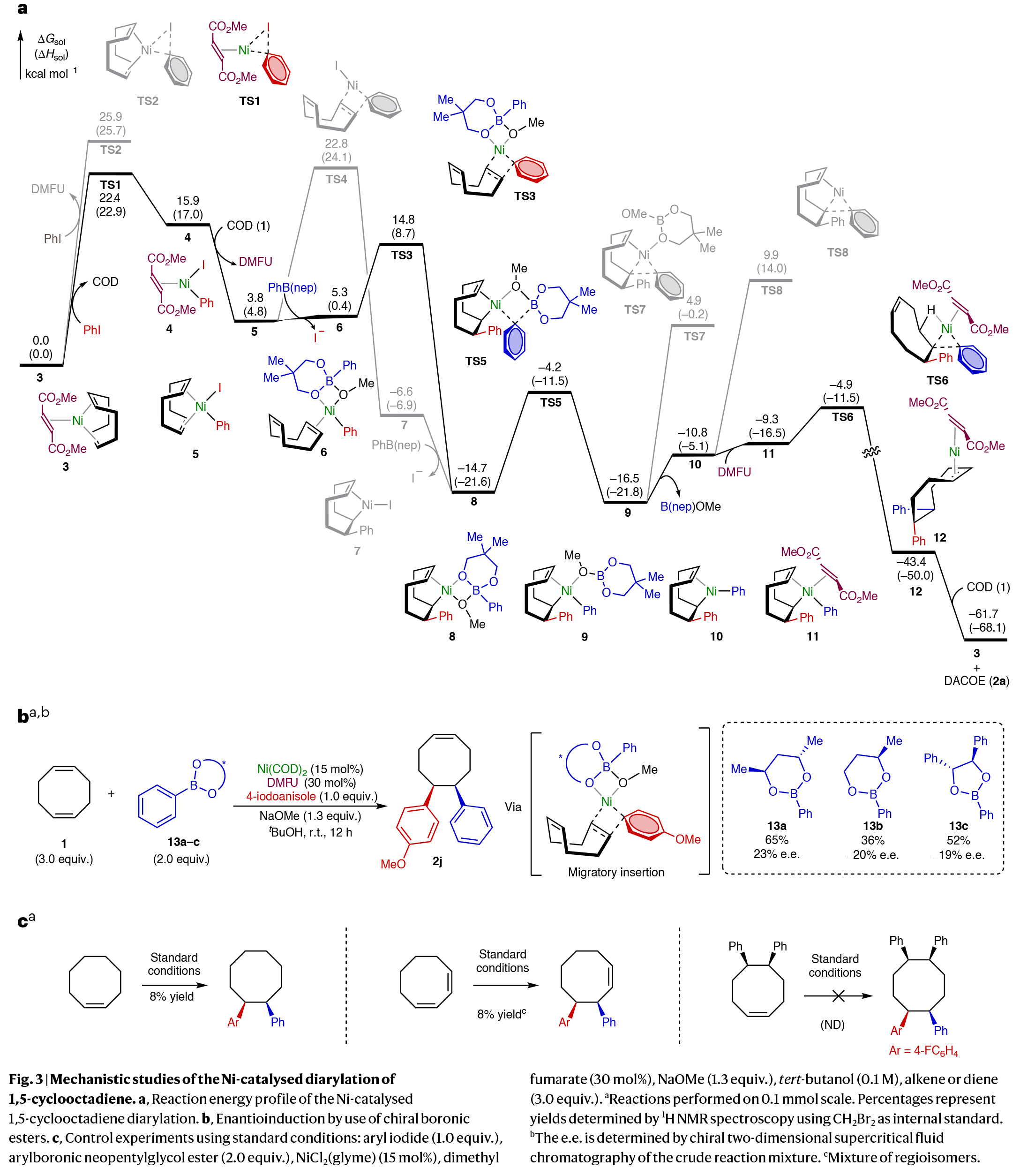
基于反应中DMFU配体的重要性和对5,6-二芳基化过程的高选择性,作者通过DFT计算对反应机理进行了考察(Fig. 3a)。实验结果表明芳基碘的初始氧化加成由DMFU配位的Ni(0)络合物促进的(通过TS1),随后进行1,2-迁移插入(TS3)到COD中的一个烯烃中。接下来,通过与芳基硼酸酯亲核试剂的转金属化(TS5),以及DMFU辅助的还原消除(TS6)得到所需产物并再生Ni(0)催化剂。有趣的是,在1,2-迁移插入步骤中既不需要第二个COD烯烃的配位,也不需要DMFU配体的配位。在该步骤中,平面四边形的芳基Ni(II)络合物与K2-配位硼酸酯阴离子结合(TS3)。在还原消除过程中,相对于其他还原消除途径(例如,TS7和TS8),强π-受体DMFU的配位显著降低了这一步骤(TS6)的过渡态能量。计算表明,1,2-迁移插入释放了COD的环张力,使该步骤是放热的。随后的转金属化和还原消除步骤的低动力学能垒表明,1,2-迁移插入是不可逆的。因此,在不对称转化中,1,2-迁移插入将决定对映选择性。作者发现1,2-迁移插入是通过与亲核硼酸酯偶联试剂(TS3)而不是与DMFU配体或碘配体(TS4)协同促进的(Fig. 3b)。此外,作者通过控制实验得出另一个烯烃部分的配位对二芳基化产物的合成至关重要(Fig. 3c)。
(图片来源:Nat. Synth.)
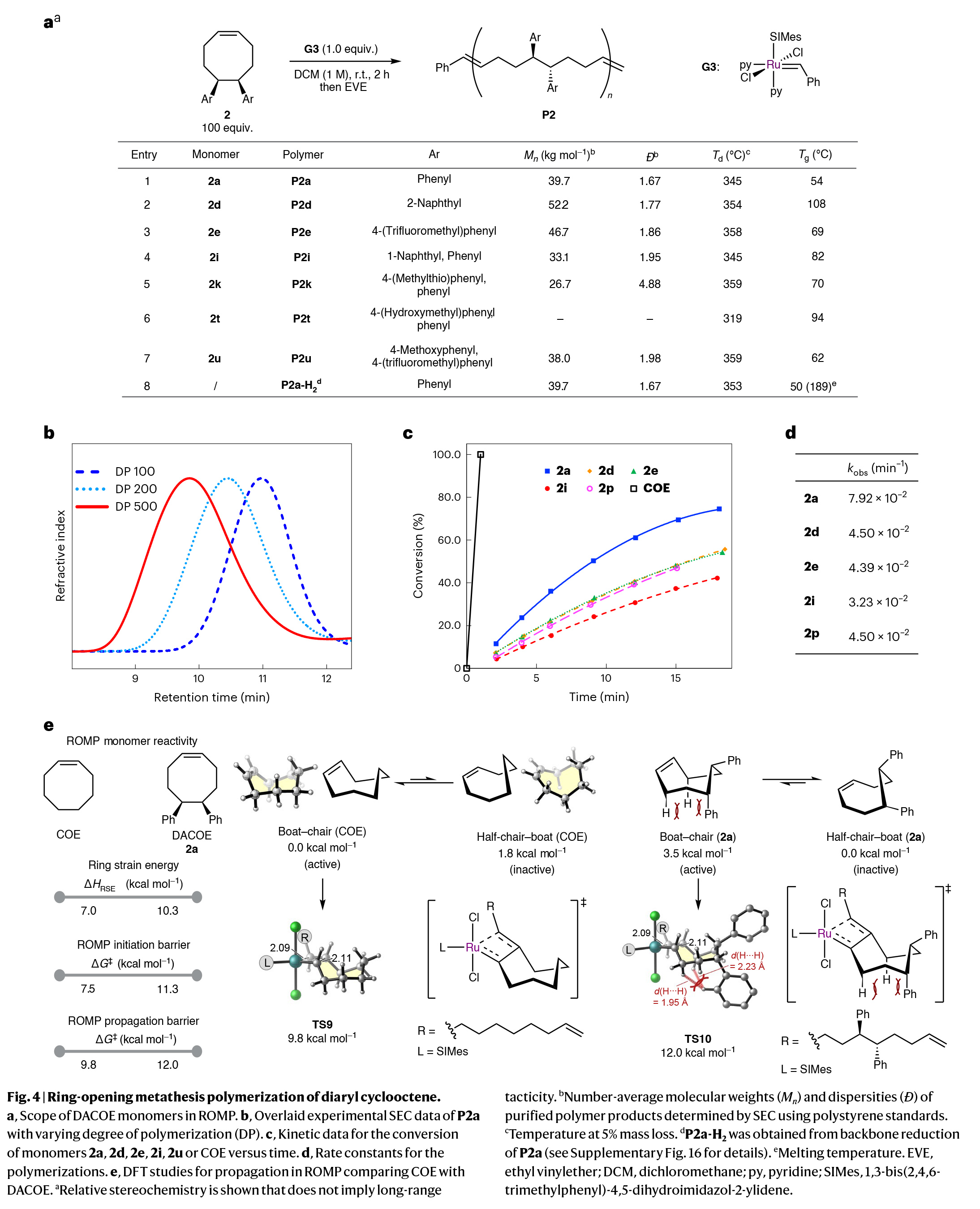
接下来,作者将注意力转向DACOE单体的聚合(Fig. 4a)。通过一系列条件筛选,作者得出当使用2 (100 equiv)和钌催化剂G3(1.0 equiv),在DCM(1 M)中室温反应2小时为最佳,可以实现高达97%的转化率。
在环辛烯体系中控制目标分子量传统上是通过链转移剂实现的。相比之下,当使用M/I比为200和500时,2a的聚合产生了可控的分子量(Fig. 4b)。虽然由于链转移反应与增殖过程同时发生,因此大分子量分布中等(Ð = 1.7-1.9),但与迄今文献中报道的传统顺式环辛烯衍生物相比,这代表了较高水平的聚合控制。
为了测试芳基的立体和电子性质对聚合速率和材料的热性能的影响,作者在优化的ROMP条件下测试了四种代表性的DACOE单体,包括带有吸电子三氟甲基的单体(2e)、推-拉电子体系(2u)和具有立体位阻的萘取代单体(2d和2i)(Fig. 4a)。实验结果表明,所有这些单体在高转化率下实现了可控的分子量和中等的分散性(Ð > 1.6)。有趣的是,与母体2a单体相比,聚合速率减慢了约50%,这表明取代基对聚合产生影响,尽管它们位于单体骨架较远的位置(Fig. 4c, 4d)。值得注意的是,无取代的COE在相同反应条件下聚合速度更快,在几秒钟内实现完全转化。
为了理解COE和DACOE单体不同聚合速率的根源,作者进行了DFT计算研究(Fig. 4e)。计算结果表明,尽管DACOE(2a)确实比COE更具张力,约为3.3千卡/摩尔,这意味着前者对ROMP有更大的热力学驱动力,但与COE相比,2a在引发和传播方面需要更高的动力学能垒。在传播阶段的速率决定性的逆-[2+2]环加成过渡态(TS9和TS10)中,DACOE采取了一种船-椅构象。这种船-椅构象对于未取代的COE来说是最稳定的,而在DACOE(2a)中,由于与苯基形成的1,3-顺位型相互作用而使这种构象不稳定。因此,2a的船-椅构象比半椅-船构象稳定性低了3.5千卡/摩尔,后者在ROMP中是无催化活性的,因为其C=C键被遮挡了。因此,相对于COE,DACOE的反应性受到苯取代基的立体效应的影响,这些基团使具催化活性的八元环船-椅构象不稳定。
(图片来源:Nat. Synth.)
总结
Keary M. Engle,Will R. Gutekunst和刘鹏课题组开发了一种镍催化COD的1,2-双碳官能化反应,从而获得结构复杂的DACOE单体。通过对这些单体的ROMP生成了之前从未被探索的立体和序列控制的聚合物结构。通过DFT计算和动力学研究阐明了两个催化过程的关键机理。此策略的发展为利用模块化多组分反应制备聚合物单体提供了新的策略,并展示了如何利用此方法来调节聚合物材料性质。
文献详情:
Ni-catalysed dicarbofunctionalization for the synthesis of sequence-encoded cyclooctene monomers,
Van T. Tran, Anne K. Ravn, Camille Z. Rubel, Mizhi Xu, Yue Fu, Ethan M. Wagner, Steven R. Wisniewski, Peng Liu,* Will R. Gutekunst,* Keary M. Engle*.
Nat. Synth., 2024
https://doi.org/10.1038/s44160-024-00618-1.


 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国