

导读
近日,日本北海道大学(Hokkaido University) Nobuya Tsuji课题组与诺奖得主、德国马克斯·普朗克煤炭研究所(Max-Planck-Institut für Kohlenforschung) Benjamin List课题组利用手性Brønsted酸——亚氨基二磷酸亚胺酯实现了对称环丙烷的包裹,并通过催化实现其开环重排,以良好的对映选择性实现了手性烯烃的合成,从而扩大了催化选择性烷烃活化的范围。计算研究表明反应中涉及环丙基正离子的参与。

(图片来源:Science)
正文
烷烃的立体选择性活化是化学领域中一个长期存在的重大挑战。虽然利用含金属的酶来氧化烷烃具有显著选择性,但化学方法却仅限于过渡金属催化的C-H官能团化反应。烷烃可以经历质子化形成五配位的碳正离子,并在强Brønsted酸的存在下可以解构成更小的碳氢化合物。然而,到目前为止,此类反应的催化立体控制版本还没有实现。近日,日本北海道大学Nobuya Tsuji课题组与德国马克斯·普朗克煤炭研究所Benjamin List课题组报道了一类手性酸,亚氨基二磷酸亚胺酯(imidodiphosphorimidates),其可以包裹住对称的环丙烷并催化其开环重排,从而以高对映选择性实现手性烯烃化合物的合成(Fig. 1)。

(图片来源:Science)
首先,作者以3-取代的双环[3,1,0]己烷1a作为模板底物对反应进行了探索,并进行了条件优化(Fig. 2)。通过一系列条件筛选作者得出当使用(S,S)-IDPi 7d (5 mol%),在MeCy(1.0 M)中,15 oC反应7天可以以84%的产率和96:4 er得到环烯烃产物2a。
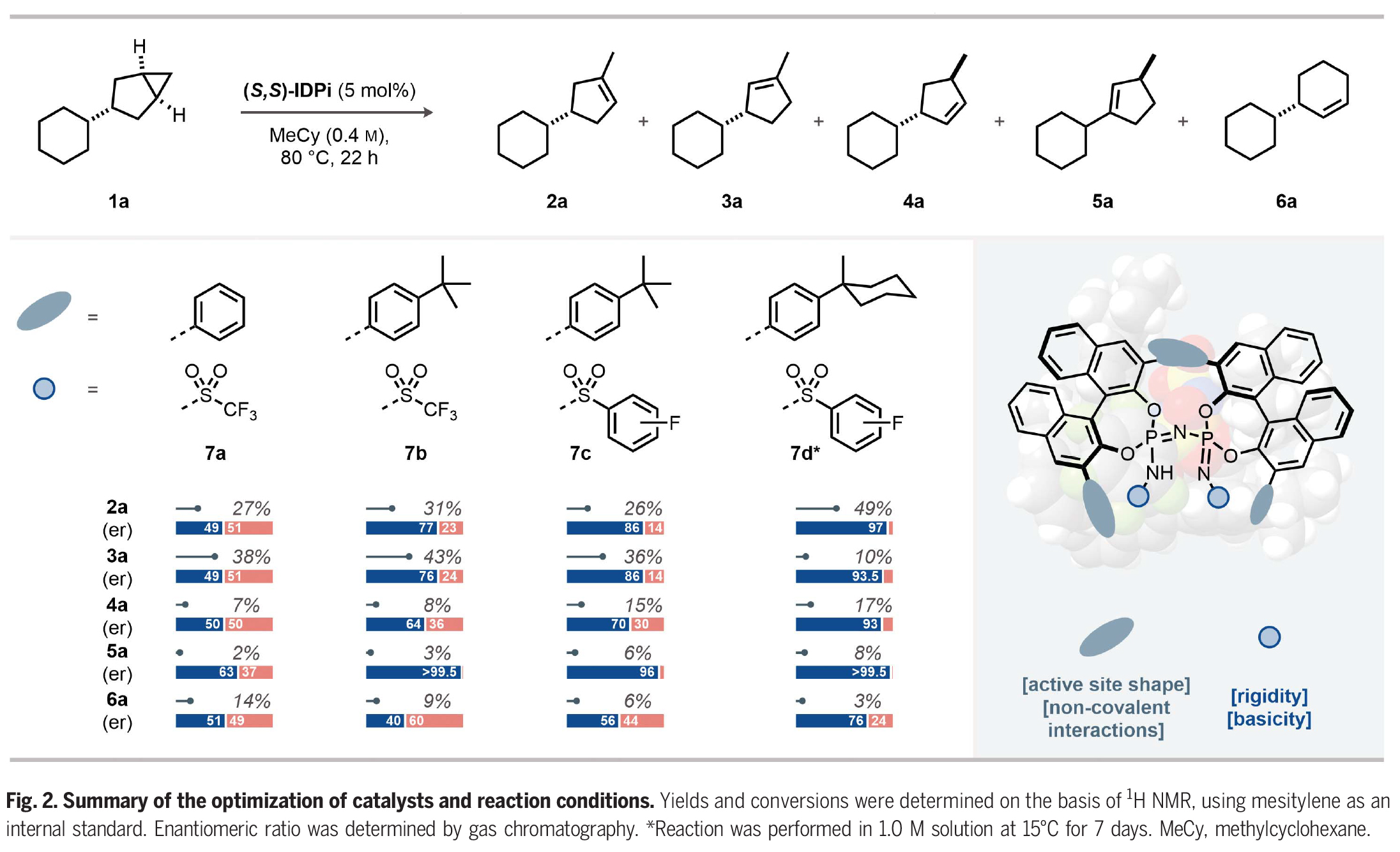
(图片来源:Science)
在得到了最优反应条件后,作者对此转化的底物兼容性进行了考察(Fig. 3)。实验结果表明,在3-取代的双环[3,1,0]己烷中,一系列环状和链状取代基均可兼容,以40-91%的产率,93:7-96.5:3.5的er得到环烯烃产物2a-2k。其中包括苄基、正丁基、甲基、苄氧基以及端烯等基团均可兼容(Fig. 3A)。此外,此方法同样可以兼容取代的双环[4,1,0]庚烷,以46-75%的产率和良好的对映选择性得到相应的环烯烃产物9a-9c(Fig. 3B)。接下来,作者探索了非并环环丙烷10的反应性。由于其具有较大的灵活性,因此将更具挑战性。事实上,IDPi催化剂7e能够在高温下活化1,2-二正丙基环丙烷(10a),产生手性二取代烯烃(11a, 12a)的混合物。其中(Z)-烯烃12a的产率为30%,对映选择性66.5:33.5 er。(E)-烯烃11a的产率为20%,对映选择性94:6 er。此外,1,2-二正丁基环丙烷(10b)也同样具有良好的兼容性,以21%的产率得到11b(95.5:4.5 er)和26%的产率得到12b(66:34 er)。作者通过将11b和12b与对映体富集的样品进行比较,确定了绝对构型(Fig. 3C)。
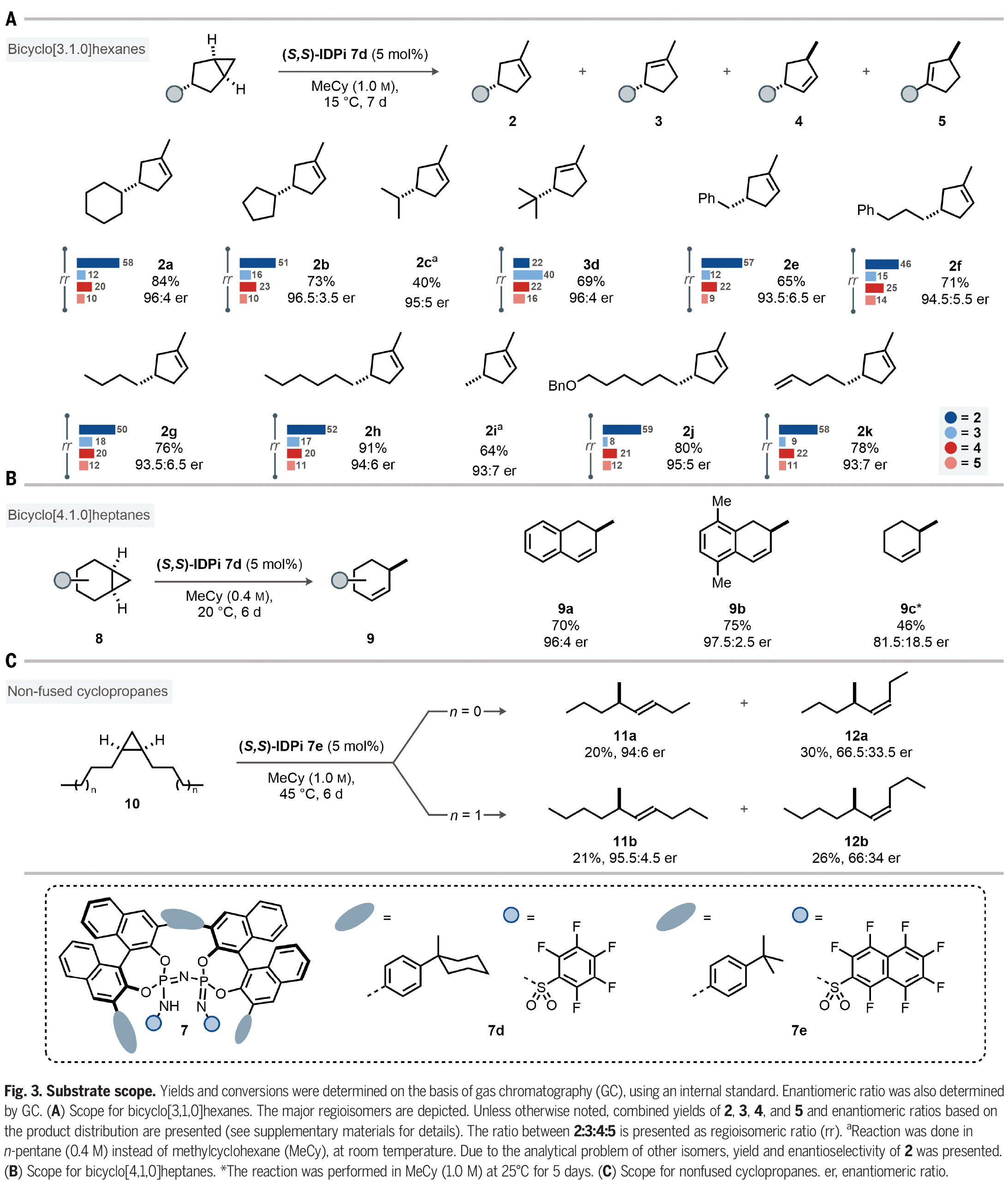
(图片来源:Science)
接下来,作者探索了此转化的反应机理(Fig. 4)。一个主要的问题是,对映体选择性是由所设计的不对称质子化过程所控制的,还是在无意的动力学拆分下驱动产物的烯烃异构化。当使用三取代烯烃2a在催化剂7d存在下60 oC反应时,作者观察到20%异构化得到3a,但2a和3a的对映体比例保持不变(Fig. 4A)。由于反应得到的3a的对映体比例低于2a,因此反应中3a是不太可能通过2a的异构化得到的。此外,双取代烯烃4h在催化剂7d存在下,即使在高温下也完全不反应。由此表明在反应条件下,4h的异构化既不能得到2h,也不能得到5h(Fig. 4B)。上述实验结果进一步得到了核磁共振动力学研究的支持。在CDCl3中,60 oC反应1天后,产物4h、5h、6h和13h达到平稳期,只有烯烃3h随着2h的消耗而持续增加(Fig. 4C)。
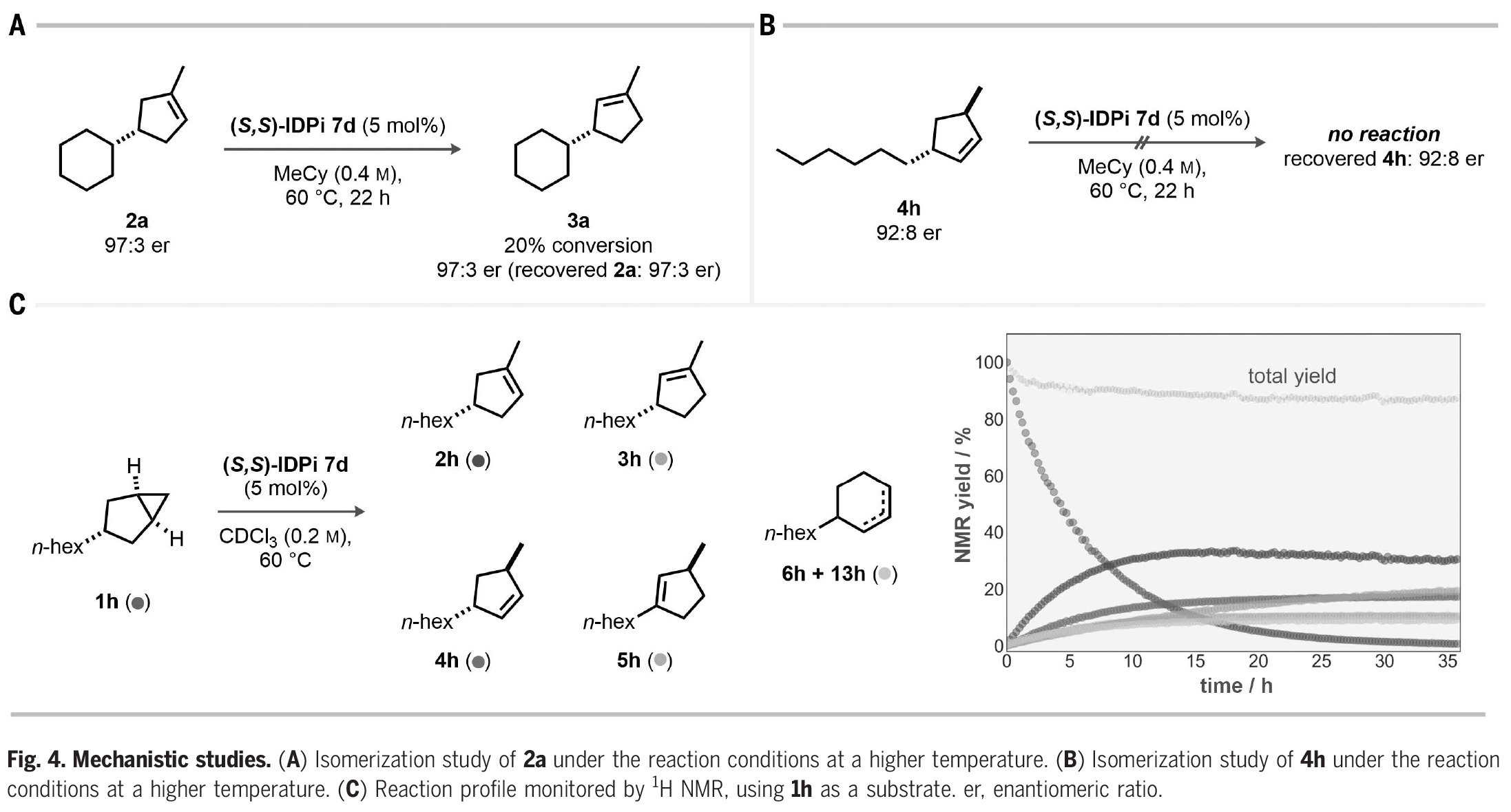
(图片来源:Science)
为了探索此转化详细的反应机理和对映选择性的来源,作者利用催化剂7d和底物1a对反应途径进行了DFT计算(Fig. 5A)。首先,底物将自身嵌入IDPi催化剂的“口袋”内,形成络合物I。在形成络合物后,底物被IDPi催化剂通过四种可能的途径质子化:通过TS1A的质子化生成碳正离子中间体II;通过TS1B的质子化生成另一个非经典碳正离子III;另一侧通过TS1A'和TS1B'的质子化生成II'和III',最终得到产物2a的次要对映体(Fig. 5B)。从Fig. 5A的键长和键角可以看出II和III的碳正离子特性。总的来说,TS4和TS3'与相应的原始对映体决定步骤的能量差为1.3 kcal/mol,这与实验结果(298.15 K时为1.7 kcal/mol)相吻合,这些中间体也可能通过重排和去质子化得到其他异构体。因此,根据异构体的不同,对映体选择性会进一步增强或降低。最后,作者对TS4和TS3'的IGMH图进行比较(Fig. 5C)。相互作用区域一般由绿色等值面组成,表明底物与BINOL取代基之间存在强烈的范德华相互作用,催化活性位点的杂原子诱导了静电相互作用。此外,阴离子中的绿色部分突出了对分子间相互作用有主要贡献的原子,表明催化活性位点中的杂原子以及脂肪族取代基通过非共价相互作用稳定底物。此外,作者对质子化步骤TS1A、TS1B、TS1A'和TS1B'也进行了研究,发现畸变和相互作用对这一步骤的选择性也起着重要的控制作用。
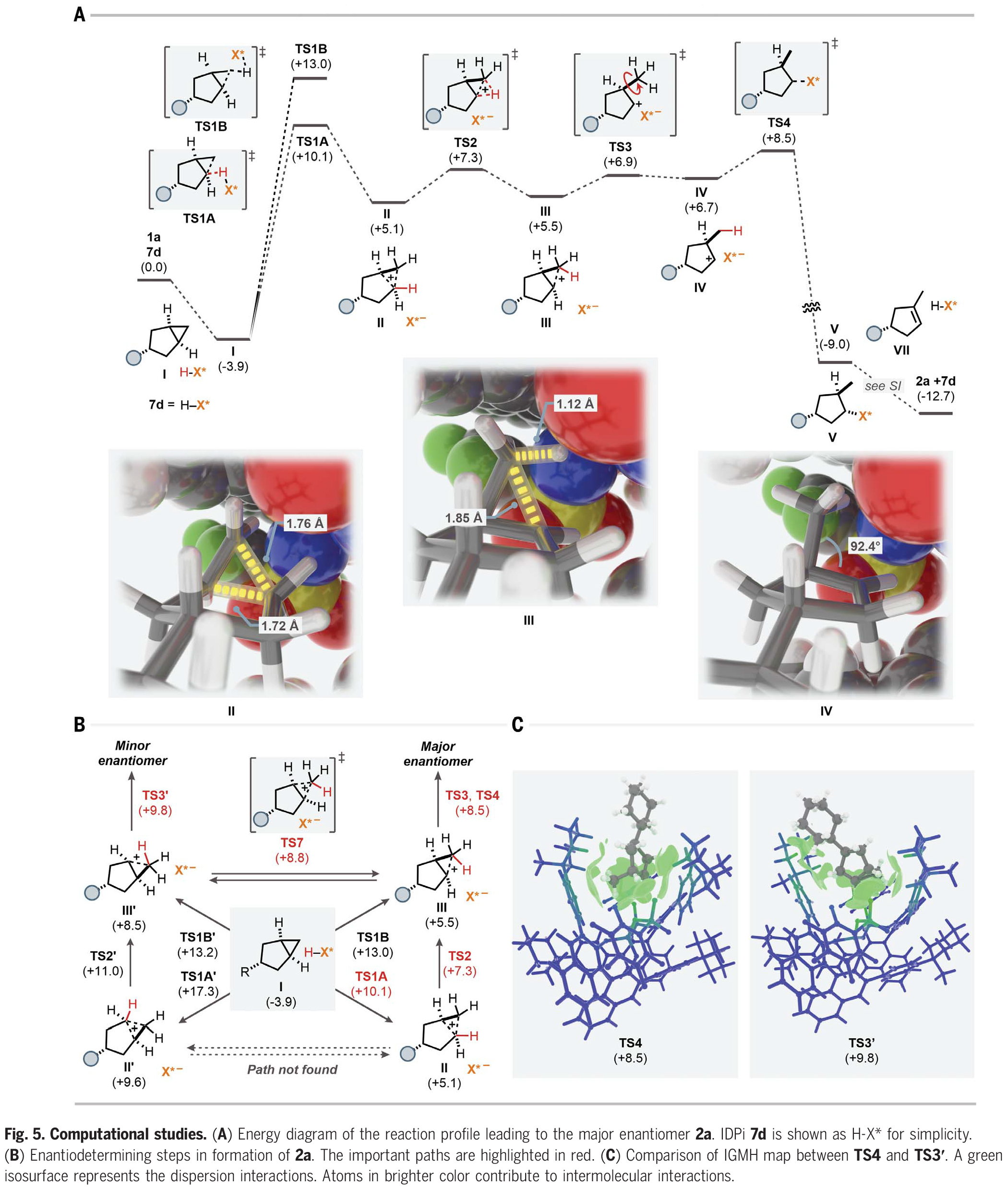
(图片来源:Science)
总结
Nobuya Tsuji课题组与Benjamin List课题组联合开发了一种利用具有特定活性位点的手性Brønsted酸催化的环丙烷的高对映选择性开环重排。一系列饱和的环丙烷均可被质子化,以优异对映选择性得到相应的烯烃。期望此方法可以突破分子立体选择性的边界,并可以进一步扩大Brønsted酸催化在烃化学中的应用。


 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国