

近日,中国科学技术大学生命科学与医学部瞿昆教授课题组、北京生命科学研究所黎斌研究员课题组,以及中国科学技术大学数学科学学院陈发来教授课题组联合完成了一项大规模研究。他们通过对百万量级单细胞多组学数据进行分析,系统评估了14种单细胞模态预测算法和18种单细胞多组学整合算法的性能。该研究成果以题为“Benchmarking algorithms for single-cell multi-omics prediction and integration”的论文,于2024年9月25日在线发表于国际知名学术期刊《Nature Methods》。

单细胞多组学技术(如CITE-seq、REAP-seq、SHARE-seq和10x Multiome等)的发展,为深入理解细胞功能和复杂的基因调控机制提供了前所未有的机遇。然而,湿实验方法通常伴随高成本、数据质量有限以及批次效应等挑战。为克服这些局限,生物信息学家基于统计模型和人工智能技术,开发了多种算法。这些算法不仅能够利用单细胞转录组数据推断同一细胞内的蛋白质丰度和染色质可及性信息,还通过将不同模态的数据映射到统一的特征空间实现数据整合,去除批次效应。这些工具大大提升了现有单细胞数据的解析能力。然而,面对海量数据和众多算法,研究人员往往难以判断哪些工具最适合他们的研究,因此,对这些算法进行基准测试(benchmarking)尤为重要。
在本次研究中,团队收集了来自47个数据集的上百万个单细胞多组学数据,涵盖多个生物样本和实验平台。他们设计了一套全面的评估流程,结合算法的准确性、鲁棒性和计算资源消耗等多维度指标,系统评估了领域内最常用的算法。结果显示,在蛋白质丰度预测方面,totalVI和scArches表现最为优异;在染色质可及性预测中,LS_Lab算法排名领先。在多组学整合分析中,Seurat、MOJITOO和scAI在垂直整合上表现突出,而totalVI和UINMF在水平整合和马赛克整合任务中展现了卓越性能。这一研究不仅为算法设计提供了新思路,还为未来多组学数据的分析和应用奠定了重要基础。为帮助科研人员选择合适的分析工具,研究团队在GitHub上发布了完整的分析流程、代码和测试数据集,供同行使用和改进。
研究团队还通过深入探讨这些算法的数学原理,发现降噪处理是提高单细胞数据预测精度的关键。在性能评估中,机器学习算法(如基于奇异值分解的LS_Lab和Guanlab-dengkw)以及基于概率模型的深度学习算法(如totalVI)均表现出显著优势。然而,研究还指出,现有模态预测算法在某些关键蛋白的预测性能上仍有待提升,染色质可及性预测的准确性也需进一步优化。
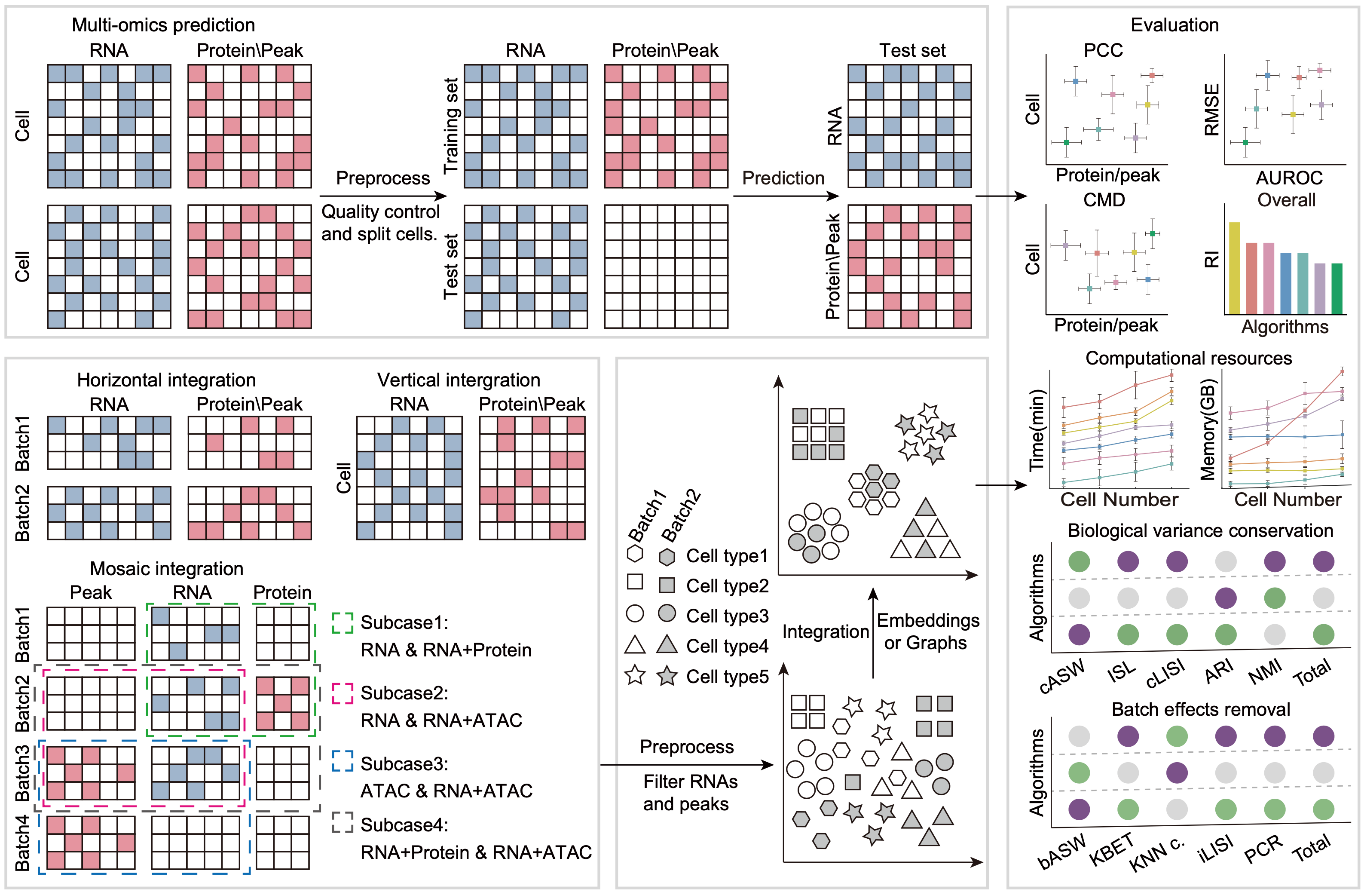
图.评估流程示意图
在组学大数据时代,对复杂数据的精确解析需要依赖生物学与数学、计算机科学的深度融合。跨学科合作不仅推动了生物医学领域的创新发展,也为未来研究提供了新的可能性。此次研究的成功正是多学科背景团队密切合作的结果,充分展示了学科交叉在现代生物学研究中的重要性。通过这样的合作,研究团队期望进一步推动单细胞多组学技术在科学研究中的广泛应用,为基础研究和临床应用提供新的洞见。



 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国