

蛋白质作为生命体的关键分子,其序列决定结构,结构决定功能,蛋白质的功能与其三维结构紧密相关。数十年来,科学家们利用 X 射线晶体学、核磁共振等技术,解析了数千种蛋白质结构,为人们理解蛋白质功能提供了重要线索。然而,面对数百万级别的蛋白质数量,解析所有蛋白质结构的任务显得异常艰巨。
**受自然语言处理领域中预训练语言模型的启发,预训练的蛋白质语言模型 (PLMs) 应运而生。**通过在海量未标记的蛋白质序列数据上进行学习,PLM 能够捕捉到蛋白质序列的复杂模式和相互作用,这为蛋白质功能的预测、结构分析以及蛋白质-蛋白质相互作用的识别带来了革命性的进步。
然而,大多数 PLM 主要关注蛋白质序列建模,忽略了结构信息的重要性,这主要是因为结构数据的缺乏。随着 AlphaFold 和 RoseTTAFold 等技术的出现,蛋白质结构预测的准确性大幅提升,研究人员也开始探索如何有效地将蛋白质结构信息整合到 PLM 中,从而训练大规模结构感知的预训练语言模型。
比如,上海交通大学自然科学研究院/物理天文学院/张江高研院/药学院洪亮教授课题组,上海交大助理研究员周冰心,联合上海人工智能实验室青年研究员谈攀,近期成功研发了一款具备结构感知能力的预训练蛋白质语言模型——ProSST。
具体来说,该模型在包含 1,880 万蛋白质结构的大型数据集上进行预训练,将蛋白质结构转化为结构化 token 序列,并与氨基酸序列一同输入至 Transformer 模型中。通过采用解耦注意力 (disentangled attention) 机制,ProSST 能够将这两类信息有效融合,从而在热稳定性预测、金属离子结合预测、蛋白质定位预测、GO 注释预测等监督学习任务中显著超越现有模型。
该研究以「ProSST: Protein Language Modeling with Quantized Structure and Disentangled Attention」为题,已入选 NeurIPS 2024。
研究亮点:
* 该研究提出了一种蛋白质结构量化器,它能够将蛋白质结构转换成一系列离散的结构元,这些离散化的结构元可有效表征蛋白质中残基的局部结构信息
* 该研究提出了一种解耦注意力机制,用于学习蛋白质氨基酸序列和三维结构之间的关系,从而促进结构离散化序列与氨基酸序列的高效信息整合
* 与 ESM 系列、SaProt 等其他蛋白质预训练大模型相比, ProSST 的参数量只有 110M,远小于经典 ESM 系列的 650M,但 ProSST 在各种蛋白质下游任务中几乎都表现出最佳性能,体现了 ProSST 模型架构设计的优越性
* 在目前最大规模零样本突变效应预测的 ProteinGym Benchmark 上,ProSST 排名第一;在最新的 ProteinGym 上,是第一个把 zero-shot 突变性能预测 Spearman 相关性做到 0.5 以上的开源模型
基于业内主流无监督预训练数据集,覆盖 1,880 万个蛋白质结构
为了实现 ProSST 的无监督预训练,研究团队主要使用了以下数据集:
* AlphaFoldDB 数据集:从超过 2.14 亿个蛋白质结构中选取了缩减 90% 的版本 (reduced version),共 1,880 万个结构,其中随机抽取 10 万个结构作为验证集,用于监测和调整训练阶段的困惑度。
* CATH43-S40 数据集:包含 31,885 个经过 40% 序列相似性去重的蛋白质晶体结构域,移除缺少关键原子(例如 Cα 和 N)的结构后,剩下 31,270 条记录,从中随机抽取 200 个结构作为验证集,用于监控和优化模型性能。
* CATH43-S40 局部结构数据集:由 CATH43-S40 数据集中提取的局部结构组成,通过构建星形图方法提取出 4,735,677 个局部结构,用于结构编码器的嵌入表征和结构码本的聚类分析。
* ProteinGYM 基准数据集:用于评估 ProSST 在零样本突变效应预测方面的能力,包含 217 个实验分析,每个分析都包含蛋白质的序列和结构信息,特别关注 66 个聚焦于热稳定性的数据集,采用斯皮尔曼系数、Top-recall 和 NDCG 作为性能评估指标。
ProSST:具有结构感知能力的 PLM,包含两个关键模块
该研究开发的 ProSST (Protein Sequence-Structure Transformer) 是一个具有结构感知能力的预训练蛋白质语言模型。如下图所示,ProSST 主要由两个模块组成:结构量化 (structure quantization) 模块和具有序列-结构解耦注意力 (sequence-structure disentangled attention) 的 Transformer 模型。
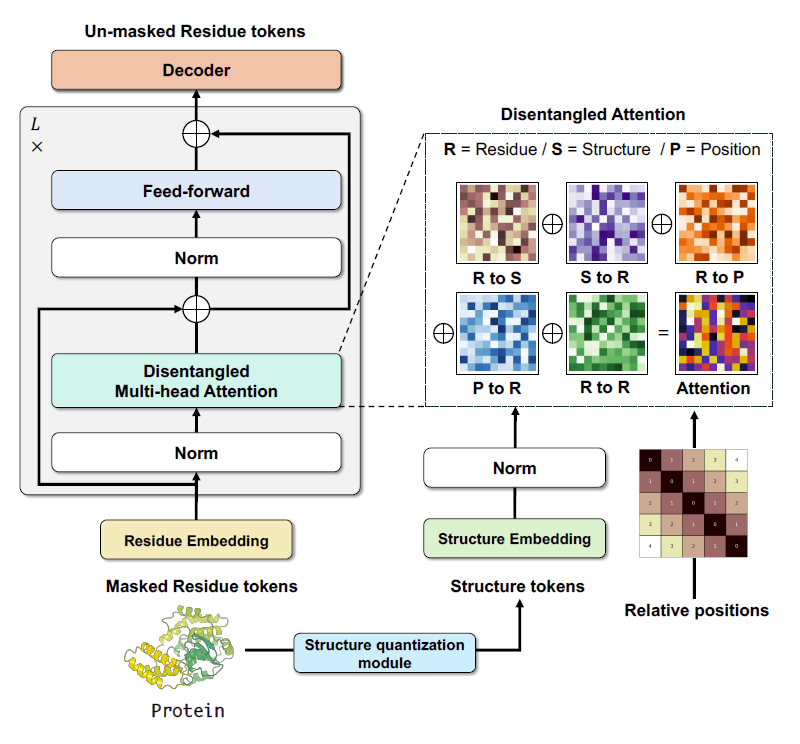
ProSST 的模型架构
结构量化模块:将蛋白质结构序列化及量化为一系列结构元
结构量化模块的目标是将蛋白质中残基的局部结构转换为离散标记。最初,局部结构 (local structure) 被预训练的结构编码器编码成了一个密集向量。随后,一个预训练的 k 均值聚类模型 (k-means clustering model) 根据编码向量为局部结构分配一个类别标签 (category label)。最后,将类别标签作为结构元 (structure token) 分配给残基。
* 相对于蛋白质整体结构,局部结构描述的颗粒度更细
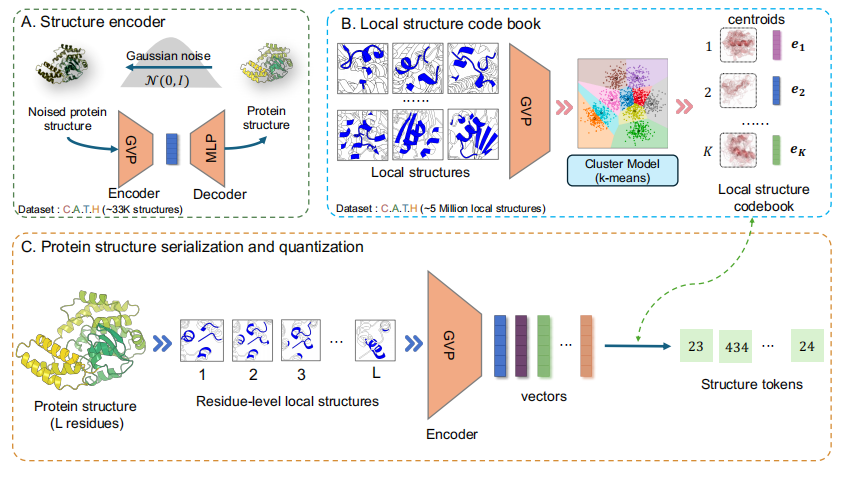
结构量化的流程
具体而言,该研究使用几何向量感知器 (GVP) 作为局部结构编码器 (local structure encoder),如下图 A 所示,该研究将 GVP 与包含位置感知的多层感知器 (MLP) 的解码器集成,形成了一个自编码器模型。整个模型使用去噪预训练目标蛋白进行训练,在 C.A.T.H 数据集上训练后,研究人员仅使用编码器的平均池化输出作为结构的最终表征。
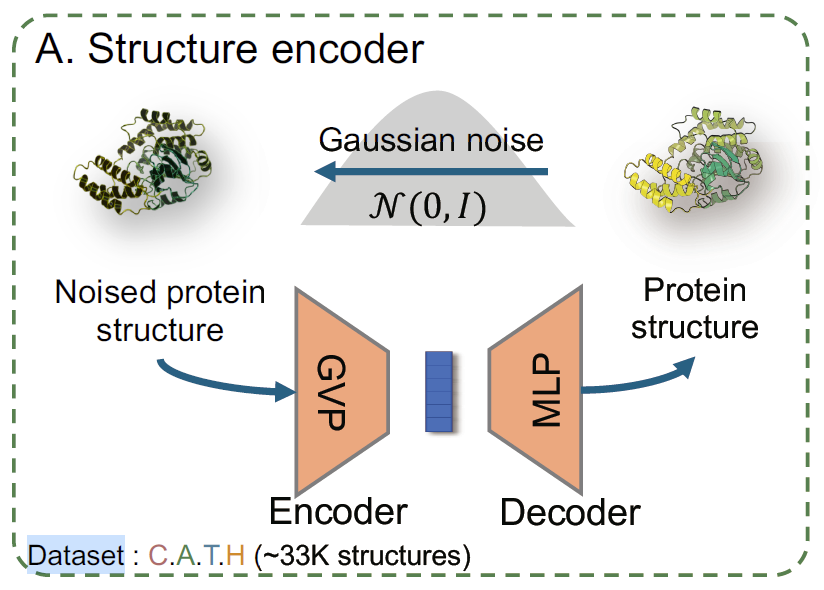
结构编码器的训练
紧接着,如下图 B 所示,该研究的结构编码器 (local structure encoder) 将表征蛋白质结构的密集向量量化为离散标记。为此,研究人员使用结构编码器 GVP 将 C.A.T.H 数据集中所有残基的局部结构 (local structures) 嵌入到连续的潜在空间中,然后应用 k 均值 (k-means) 算法在这个潜在空间中识别 K 个质心 (centroids),这些质心构成了结构码本 (structure codebook)。
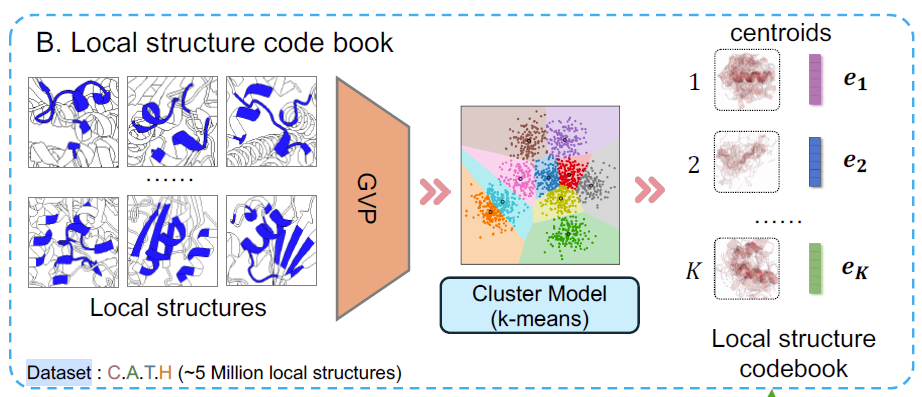
局部结构聚类和标记
最后,对于蛋白质序列中位置 i 的残基,该研究首先基于其局部结构构建图 Gi,然后使用结构编码器 GVP 将其嵌入到连续向量 (vector) ri 中。总体而言,如下图 C 所示,整个蛋白质结构可以序列化并量化 (serialized and quantized) 为一系列结构元 (structure tokens)。
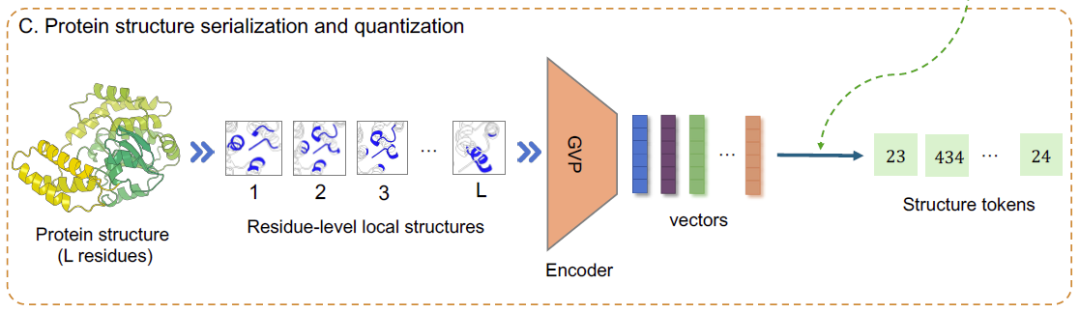
将蛋白质结构转化为结构元序列
序列-结构解耦注意力:使模型学习残基与残基、残基与结构之间的关系
该研究受到了 DeBerta 模型的启发,旨在通过解耦注意力的方式来学习残基序列(氨基酸序列)和结构序列,以及相对位置三者间的相互关系,从而使得模型能够处理蛋白质序列和结构信息,并通过解耦的方式来提高模型的性能和稳定性。
具体来说,对于蛋白质一级序列中第 i 个残基,可以通过 3 项来表示:Ri 表示氨基酸序列 token 的编码,Si 代表氨基酸的局部结构 token 编码,而 Pi|j 表示第 i 个残基在位置 j 的 token 编码。如下图所示,该研究的序列-结构解耦注意力机制包括残基对残基 (R to R)、残基对结构 (R to S)、残基对位置 (R to P)、结构对残基 (S to R)、位置对残基 (P to R) 这 5 种类型,使得模型能够更细致地捕捉蛋白质序列和结构之间的复杂关系。

ProSST 的模型架构
ProSST 性能全面领先,结构信息的纳入大幅提高模型表征能力
为了验证 ProSST 在零样本突变效应预测 (zero-shot mutant effective prediction) 的有效性,该研究将其与多种顶尖模型进行了比较,涵盖序列模型 (sequence-based models)、结构-序列模型 (structure-sequence model)、逆向折叠模型 (inverse folding models)、进化模型 (evolutionary models) 和集成模型 (ensemble models)。
如下表所示, 在 ProteinGYM 的基准评测下,ProSST 的表现优于所有对比模型,并在稳定性 (stability) 上达到最佳。此外,ProSST (-structure) 表现与其他序列模型相当,这证实了 ProSST 的性能提升主要归功于其对结构信息的有效整合。
* ProSST (-structure) 没有纳入结构信息模块
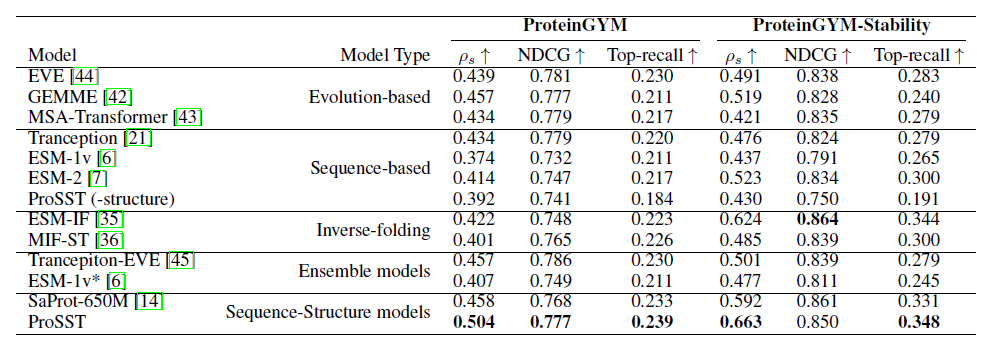
ProSST 与其他模型的零样本突变预测性能比较
对于监督学习,该研究选择了热稳定性预测 (Thermostability)、金属离子结合预测 (Metal Ion Binding)、蛋白质定位预测 (DeepLoc) 以及 GO 注释预测 (MF/BP/CC) 四大蛋白质下游任务,并且将 ProSST 与 ESM-2、ESM-1b、SaProt、MIF-ST、GearNet 等其他蛋白质语言模型进行比较。结果如下表 2 所示,ProSST 在所有模型中获得最佳结果,在所有 6 种设置中获得了 5 个第一名和 1 个第二名。
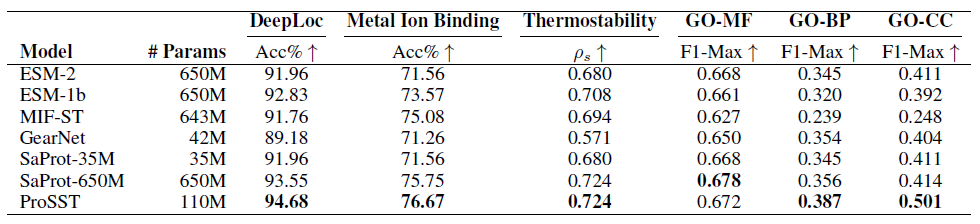
下游任务的监督微调比较
蛋白质语言模型:连接大数据与生命科学的桥梁
自 ChatGPT 等大语言模型发布以来,基于大规模蛋白质序列的预训练模型 (PLM) 已成为生命科学领域的热门研究。目前,PLM 研究主要分为两个方向:
* 检索增强型 PLM:这类模型会在训练或预测阶段整合多序列比对 (MSA) 信息,如 MSATransformer 和 Tranception,以提升预测性能。
* 多模态 PLM:与仅使用序列信息的模型不同,多模态 PLM 整合蛋白质结构等额外信息,如本文所述的 ProSST 模型,通过结构 token 序列与氨基酸序列的融合,即可增强模型的表征能力。
在检索增强型 PLM 方面,今年 4 月,复旦大学等机构的研究团队推出了 PLMSearch,这是一种基于序列输入的同源蛋白质搜索方法。该研究可利用预训练的蛋白质语言模型获取深层表征,并预测结构相似度。相关研究已发表于 Nature Communication。
在多模态 PLM 方面,浙江大学陈华钧教授团队前不久提出了一种针对蛋白质优化的全新去噪蛋白质语言模型 (DePLM),该模型通过优化进化信息,可提高蛋白质优化任务的性能,相关成果已成功入选顶会 NeurIPS 24。
随着这些突破性研究的不断涌现,PLM 正逐渐成为探索生命科学未知领域的强大工具,在蛋白质功能预测、相互作用预测及表型关联预测等领域具有巨大潜力,有望为疾病治疗和改善人类生活提供新思路。
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国