


婴儿出生后,会迅速定植来自母亲和环境中的微生物,儿童时期的疾病也可能会受到婴儿期肠道定植微生物的影响。然而,对生命早期肠道菌群的研究大多受限于16S分析的分辨率和较小的样本量,且对新生儿期(≤1月龄)的研究不足。
近期,Nature发表了迄今为止最大规模的新生儿肠道菌群的宏基因组学研究,该研究来自英国惠康桑格研究所(Wellcome Sanger Institute)的Trevor Lawley团队,研究结果显示:
分娩方式是影响新生儿期肠道菌群组成的最主要因素,剖宫产会造成新生儿肠道菌群发育不良,增加医院环境相关机会致病菌的定植,破坏母婴间的拟杆菌传递,但这些变化是否对长期健康有影响还需进一步研究。
该研究选择了婴儿生物群落研究(BBS)中的596名健康且足月生产的婴儿,其中314名是阴道分娩,282名是剖宫产分娩。研究人员分别收集婴儿出生后第4、7和21天的粪便样本(每个婴儿至少包含其中一个样本),同时采集其中178名产妇的粪便样本(仅在分娩前后或分娩过程中采集一次)。研究人员对采集到的1679份粪便样本进行了宏基因组测序及分析。
首先,他们发现,在婴儿出生后的1个月内,肠道微生物呈高度动态化(图1),且随着年龄的增长,菌群种类也逐渐增加。利用置换多元方差分析(PERMANOVA)发现,个体差异大约能解释57%的不同肠道微生物组成。
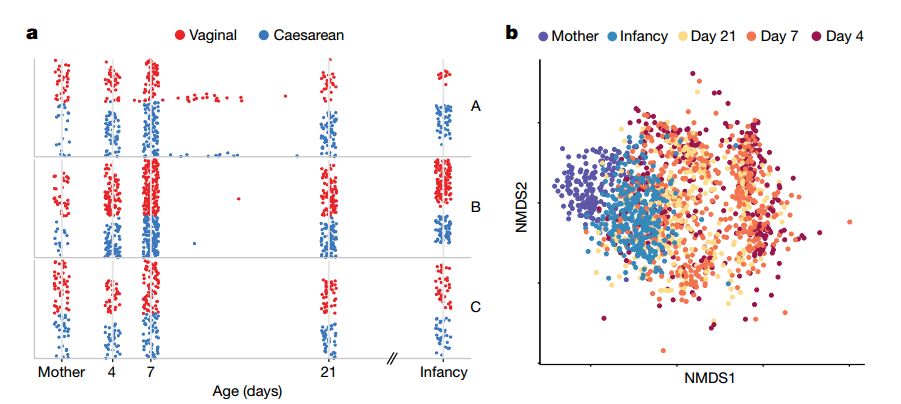
图1 新生儿肠道微生物是高度动态化的
接下来,研究人员进一步运用横断面PERMANOVA分析了临床因素对新生儿肠道菌群构成的影响(图2a),临床因素包括分娩方式、是否母乳喂养、在何种医院出生、是否接受抗菌药物治疗等等,发现分娩方式是影响新生儿肠道菌群构成的最主要因素。
不同分娩方式到底会对婴儿肠道菌群造成怎样的影响呢?
经过分析发现,经阴道分娩的婴儿肠道内双歧杆菌属(长双歧杆菌、短双歧杆菌)、埃希氏菌属(大肠杆菌)、拟杆菌属(普通拟杆菌)和副拟杆菌属(狄氏副拟杆菌)丰度很高,占据了肠道微生物的68.3%(图2b左)。而这类菌群在剖宫产的婴儿肠道中则很少,主要是屎肠球菌、表皮葡萄球菌、副血链球菌、产酸克雷伯氏菌、肺炎克雷伯氏菌、阴沟肠杆菌和产气荚膜梭菌,这些菌都与医院环境和早产相关。出生后第4天,这些菌属占到婴儿总肠道微生物的68.25%(图2b右)。
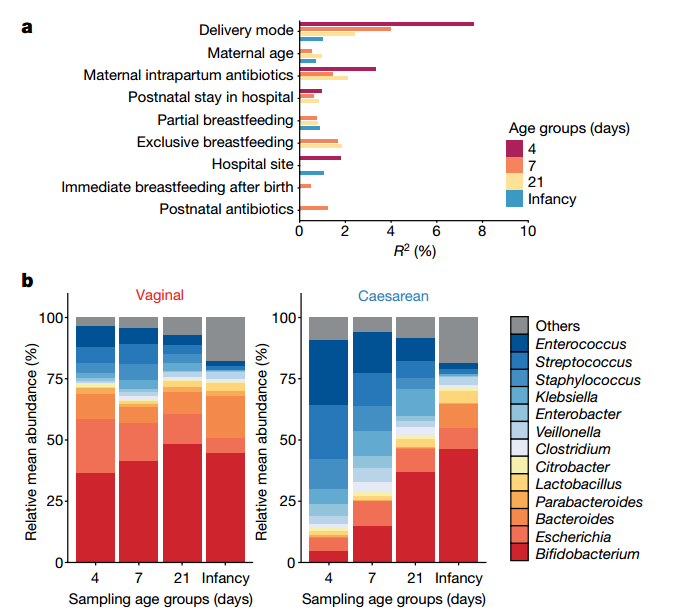
图2 a. 分娩方式是影响新生儿肠道菌群构成的最主要因素;
b. 经阴道分娩(左)和剖宫产(右)婴儿在出生后第4、7和21天时的肠道微生物构成
对178对婴儿和母亲肠道菌群的测序分析发现,经阴道分娩的婴儿74.39%的肠道微生物来源于母亲肠道微生物,而剖宫产婴儿只有12.56%。特别值得注意的是,若没有从母亲处获得拟杆菌属的普通拟杆菌(B. vulgatus),那么在以后很长一段婴幼儿时期都很难再获得(图3)。这些结果提示,宝宝出生前后是获得菌群的最佳时机。
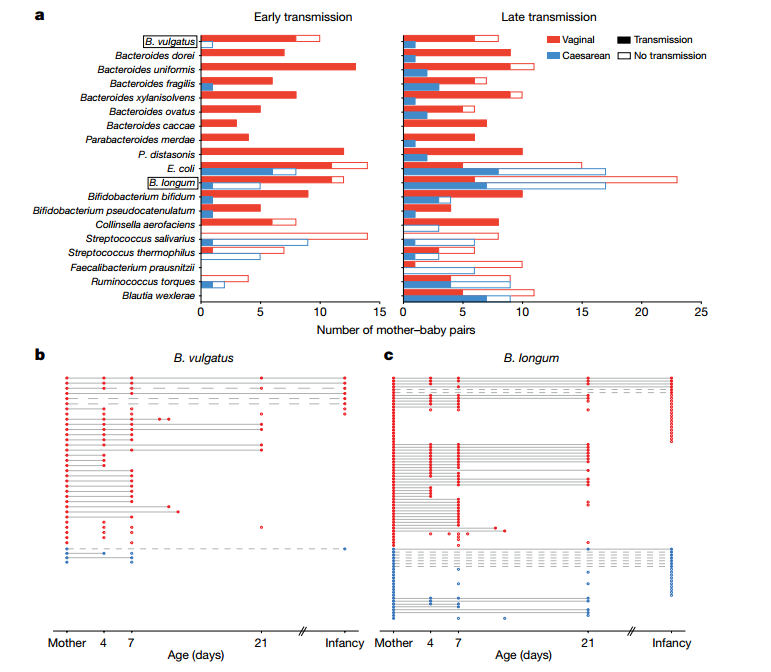
图3 经剖腹产分娩的婴儿出现母体微生物株的传播中断
作者发现,83.7%的剖宫产宝宝肠道微生物中出现了在医院广泛存在的机会致病菌,而经阴道分娩宝宝只有49.4%(图4)。从出生到21天,这些病原菌所占的菌群种类占剖宫产婴儿肠道菌群的30.4%,在经阴道分娩婴儿中只占9.8%。研究人员对这些病原菌进行了分离和培养,发现这些菌携带毒力因子和抗生素耐药相关基因,可能增加感染风险。
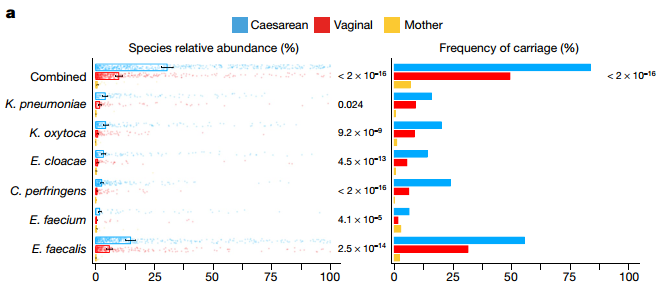
图4 超过80%的剖宫产婴儿携带医院中广泛存在的机会性致病菌
虽然剖宫产的确改变了新生儿的肠道菌群,但尚不了解这些改变是否会影响长期健康。接下来随着母婴喂养、饮食结构变化和年龄增长,这种差异可能会逐渐淡化,但无疑是需要关注的又一个重要问题。
可借鉴之处
1
首次对新生儿期(≤1月龄)的肠道菌群进行了高分辨率宏基因组学研究,揭示了分娩方式是影响新生儿期肠道菌群组成的重要因素。
2
进一步分析不同时间段的婴儿菌群,揭示菌群差异随时间减少。通过对母婴配对的创新性分析发现,虽然差异随时间减少,但剖宫产婴儿持续缺少母亲传递的拟杆菌,并在以后很长时间内难以获得。
3
有别于以往对于肠道菌群的常规分析,该研究通过分离培养肠道菌群,分析其携带的毒力因子和抗生素耐药基因,为肠道菌群与人体健康之间关系的研究提供了新视角。
局限性
该研究仅揭示剖宫产会影响新生儿期的肠道菌群构成,造成致病菌定植,但生命早期肠道菌群紊乱以及携带致病菌的临床后果有待进一步研究。只有继续跟进,观察这些婴儿的成长过程,才能知道新生儿时期微生物构成的不同是否真的会影响长期健康,未来我们该如何改善这一情况。
本期关键词
16S rDNA测序和宏基因组测序
16S rDNA基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,位于原核细胞核糖体小亚基上,包括 10 个保守区域和 9 个高变区域,高变区具有属或种的特异性。
因此16S rDNA可以作为揭示生物物种的特征核酸序列,被认为是最适于细菌系统发育和分类鉴定的指标。由于16s rDNA较长(1.5kb),考虑到测序平台的读长限制,一般只能对可变区进行测序。16s rDNA包含有9个可变区,分别是v1-v9。一般对v3,v3-v4和v6可变区域进行扩增和测序。
宏基因组测序直接从环境样品中提取全部微生物DNA,构建宏基因组文库,利用高通量测序技术进行全部微生物基因组的序列测定。
区别于16s rDNA测序,通过宏基因组测序不仅可以分析菌群组成,还可以进行功能基因的发掘,分析微生物群体基因组成及功能,解读微生物群体的多样性与丰度,发掘和研究新的、具有特定功能的基因。
目前宏基因组测序技术为微生物的研究和发展提供了很好的策略,在发现新基因,开发新的微生物活性物质,研究微生物种群结构、基因功能活性、微生物之间的相互协作关系以及微生物与环境之间的关系等方面得到广泛的应用。
参考文献
Yan Shao, Samuel C. Forster, Evdokia Tsaliki, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth [J]. Nature, 2019,574:117-121.
本期嘉宾

贾鑫淼
博士,生物信息学,北京协和医院-中心实验室-统计与生信平台。
北京协和医院-中心实验室-统计与生信平台基于循证医学、医学统计学、临床流行病学、生物信息学理论与前沿技术,在院内外开展方法学咨询服务与大数据分析技术支持。从疾病的病因、诊断、治疗、预后多角度开展临床研究,为疾病预防、早期干预、新药和新治疗方法的临床应用提供科学的证据支持,促进转化医学发展与科研成果转化。
欢迎有兴趣的同道与我们联系,联系方式:pumchstat@126.com。
栏目策划

吴志宏教授
北京协和医院骨科教授、博导、中心实验室副主任、实验动物管理委员会主任、骨骼畸形的遗传学研究北京市重点实验室副主任、北京市生物医学工程高精尖中心学术委员会委员、医工整合联盟副理事长、中华医学会骨科分会基础学组委员。
(本文图表来自于英文原文)
版权声明:
协和医学杂志倡导尊重和保护知识产权。欢迎转载、引用,但需取得本平台授权。如您对文章内容版权存疑,请发送邮件medj@pumch.cn,我们会与您及时沟通处理。本站内容及图片仅供参考、学习使用,不为盈利且不作为诊断、医疗根据。

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国