

芳香化酶抑制剂能特异性导致芳香化酶失活, 阻断芳构化反应, 抑制雌激素生成,降低血液中雌激素水平从而达到治疗乳腺癌的目的。多用于抗雌激素(他莫昔芬)治疗失败的绝经后晚期乳腺癌患者。常用的芳香化酶抑制剂:依西美坦、来曲唑、阿那曲唑。
芳香化酶(aromatase , AR) (也叫雌激素合成酶) )是微粒体细胞色素P450 的一种复合酶, 它由血红蛋白P450a rom和还原型辅酶NADPH 组成, 广泛存在于卵巢、肝脏、肌肉、脂肪和乳腺癌细胞中,是催化生物体内雄激素向雌激素转化的关键酶和限速酶,可将雄激素的A环芳香化, 脱去19 位的碳原子并将1位的羰基转化为羟基, 这样就将催化雄烯二酮和睾酮等雄激素转化为雌酮和雌二醇, 后两者即为绝经后女性雌激素的主要来源。雌激素与肿瘤进展有关,芳香化酶在雌激素生物合成中起最终的限速催化作用。
芳香化酶抑制剂(aromataseinhibitor,AI)能特异性导致芳香化酶失活, 阻断芳构化反应, 抑制雌激素生成,降低血液中雌激素水平从而达到治疗乳腺癌的目的。多用于抗雌激素(他莫昔芬)治疗失败的绝经后晚期乳腺癌患者。常用的芳香化酶抑制剂:依西美坦、来曲唑、阿那曲唑。 1
1.芳香化酶的分布与调节雌激素可在男性和女性的各种组织中合成。绝经后妇女循环雌激素水平主要依靠脂肪组织合成的雌激素来维持, 然而人们发现绝经后妇女乳腺组织的雌二醇水平比血浆中高10倍。有报道指出, 芳香酶的活性以及芳香化酶的mRNA在正常乳腺组织和乳腺肿瘤中都存在。某些临床观察显示: 肿瘤内的芳香化与肿瘤对芳香化酶抑制剂治疗后雌激素合成受抑的反应相关, 局部雌激素的产生对肿瘤的增生可能也有重要作用。然而, 在人体乳腺癌组织匀浆中测得的芳香化酶活性相对较低, 而且不足以催化形成足量的雌激素去激活雌激素受体。另有研究表明, 局部雌激素浓度足以刺激肿瘤生长。同时组织培养发现, 某些由雌激素刺激的肿瘤也可以通过睾丸酮加速增生, 且该刺激作用可以通过芳香化酶抑制剂阻滞。提示睾丸酮经芳香化生成雌激素。由此可见, 肿瘤中的芳香化酶对刺激肿瘤增生的雌激素的生成具有重要意义。
- 芳香化酶抑制剂的分类研究表明:有三分之二的乳腺癌是雌激素依赖性的, 它们能在雌激素减少后得以消退。1973年, Schwarsel 首次提出芳构酶抑制剂能特异性导致靶酶失活, 阻断芳构化反应, 抑制雌激素生成,降低血液中雌激素水平从而达到治疗乳腺癌的目的, 并相继报道了一系列抑制AR 活性的化合物。芳构酶抑制剂作为乳腺癌治疗的一种全新思路引起了相关学者的关注, 经近三十年的努力, 已开发上市了三代AR 抑制剂, 作为治疗乳腺癌的二线或三线药物, 在美国、日本等已广泛用于临床。AR 抑制剂按其结构可分为甾体和非甾体两类。具体分类见表1,具体结构见图1.

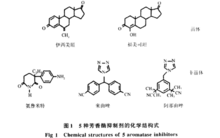
2.1 甾体类芳香化酶抑制剂
2.1.1依西美坦(exemestane,Aromasin)
1988 年, 意大利Pharmaela &Upjoin公司研究开发成功新一代抗芳香化酶药物依西美坦,属于第三代芳香酶抑制剂 并于1999 年底获得欧美等国家批准, 主要用于治疗绝经后妇女的晚期乳腺癌。
依西美坦特点:①与体内芳香化酶的结合是不可逆的, 实属芳香化酶灭活剂。依西美坦为芳香化酶的假性底物, 其结构与酶的自然底物雄烯二酮和睾酮相似, 可通过与酶的活性位点不可逆地结合而使其失活(该作用也称“ 自毁性抑制”), 从而明显降低绝经妇女血液循环中的雌激素水平。②对芳香化酶的灭活作用无“反弹” 。体外实验发现, 当从细胞培养液中分别除去依西美坦、氨基导眠能和阿那曲唑后, 依西美坦组JEG-3 细胞中的芳香化酶仍处于受抑状态, 而后两组JEG-3细胞的芳香化酶活性反较原来有所提高。应用芳香化酶抑制剂后所致的酶活性提高可能会损害芳香化酶抑制剂的长期疗效, 而依西美坦具有的持续抑制芳香化酶活性且不提高芳香化酶蛋白水平的性质, 则有益于减少这种风险。③底物选择性高, 对肾上腺内皮质类固醇和醛固酮的生物合成无明显影响。依西美坦即使在高于其抑制芳香化酶作用浓度600倍时, 对类固醇生成途径中的其他酶也无明显影响。
2002 年5月, 各国际协作组合作, 选择绝经后乳腺癌患者, 开展了42 项依西美坦临床试验, 内容涉及乳腺癌的预防、术后辅助治疗、新辅助治疗、转移性乳腺癌的治疗等方面, 以期取得充分的循证医学依据。
2.1.2福美司坦(formestane)
福美司坦为人胎盘微粒体、大鼠卵巢微粒体、人乳腺癌细胞和乳腺癌活检样品中的芳香化酶抑制剂,属于第二代芳香化酶抑制剂。徐积恩的研究显示,福美司坦可引起大鼠卵巢和人胎盘微粒体的迅速抑制,给药后3~20 min 活性抑制可达50%。福美司坦50mg/(kg·d)后采用同位素示踪法测出雄烯二酮转化为雌激素的抑制率达90%。
Dowsett M等的研究显示,给予绝经后女性福美司坦500 mg,24 h 内平均血浆雌激素水平下降40%,给药7 d 可下降80%;每2 周注射250 mg,8周内抑制雌激素水平约60%;其口服最大有效剂量为250 mg/d。由于绝经前女性雌激素生成主要在脑垂体释放的促性腺激素控制下的卵巢,不受芳香化酶抑制作用而减少,因而绝经前女性单用福美司坦对雌二醇水平无明显影响,临床表现为产生拮抗作用。
Brodie AM等的研究采用二甲基苯并蒽(DMBA)诱发大鼠乳腺癌模型,给予福美司坦50 mg/kg,im,bid,给药4 周后,约90%的大鼠肿瘤缩小程度>50%,4%的大鼠肿瘤完全消退。Chander SK等的研究显示,福美司坦12.5 mg/kg+5%柠檬(limonene)对致癌物N-亚硝基脲(NMU)诱发的大鼠乳腺肿瘤有效,抑制率为83.3%,优于福美司坦单独使用。Dowsett M等发现,采用口服/非胃肠道吸收的所有途径给药,福美司坦对血清中的雄烷二醇、睾丸素或5α-二氢睾丸酮水平没有影响;福美司坦250 mg/d,po,给药4 次后,34%的患者出现性激素结合球蛋白(SHBG)水平下降,采用气-质联用(GC-MS)分析方法测定非胃肠道吸收给药对血清雌酮水平的抑制约为40%。福美司坦对雌激素生物合成的抑制并不改变绝经后女性的雌激素前体含量。
福美司坦的新用途和用法逐渐被发现,比如对早期子宫内膜癌的治疗、对念珠菌的抑制作用、通过与曲妥珠单抗联用治疗乳腺癌、与宽缨酮的联合抑制芳香化酶作用,增强特异性抗体对肿瘤细胞的抑制作用、降低单一用药的不良反应等。
2. 2 非甾体芳香化酶抑制剂
2.2.1氨鲁米特(aminoglutethimide,AG,氨基导眠能,氨格鲁米特,氨苯哌酮)
氨鲁米特是一种类固醇生物合成抑制剂。氨鲁米特虽是芳香化酶的强效抑制剂, 但对转化类固醇成为孕烯醇酮的碳链裂解酶也有抑制作用, 减少了皮质类固醇的体内合成, 常引起皮疹、共济失调和磕睡, 发生率高达60 %, 需与氢化可的松合用。偶可出现白细胞减少,血小板减少和甲状腺功能减退。非甾体芳香化酶抑制剂含有杂原子(通常带有含N 的杂环部分)。一般通过与细胞色素P 450的血红素铁结合, 干扰甾体经基化过程。如同多数甾体类抑制剂一样,这些化合物是可逆性芳香化酶抑制剂。总体上看, 多数可逆性非甾体芳香酶抑制剂特异性较低, 会不同程度地抑制其它细胞色素P 450 参与的甾体合成的羟基化反应。除氨鲁米特外, 其它非甾体芳香酶抑制剂对芳香化酶具有较高的选择性。
2.2.2来曲唑( letrozole)
来曲唑是高选择性、第三代芳香化酶抑制剂。它在疗效、安全性以及经济学上的优越性已经为多项临床研究所证实。来曲唑作为可逆性结合的芳香化酶抑制剂, 通过抑制外周和肿瘤组织中的芳香化酶, 有效降低血浆雌激素水平, 从而去除对激素敏感肿瘤的刺激。约有1/3以上的乳腺癌依赖于雌激素的刺激而继续发展。在绝经前妇女的雌激素主要来源于卵巢,而绝经后妇女雌激素则主要由肾上腺、脂肪、肌肉、肝脏产生的雄激素经芳香化酶转化而来。因而对绝经后妇女而言, 通过抑制芳香化酶, 即可减少体内雌激素产生, 从而达到治疗乳腺癌的目的。类固醇分子的芳香化是雌激素生物合成过程中的最后步骤, 选择性芳香化酶抵制剂是能够作用于雌激素生物合成最终阶段的内分泌治疗药物, 不干扰其他固醇类激素的合成, 所以能更好地针对目标, 在作用和疗效上有更好的选择性。
绝经后晚期乳腺癌的一线治疗:PO25是1项多中心、随机、双盲、双模拟对照试验, 旨在比较第三代芳香化酶抑制剂和他莫昔芬作为绝经后乳腺癌一线治疗的可行性。此研究涉及29个国家的10个医疗研究机构, 共有916位绝经后晚期乳腺癌入组试验, 中位随访期为32个月。结果来曲唑组的疗效均优于他莫昔芬组。
二线解救治疗:来曲唑与第一代芳香化酶抑制剂氨基导眠能(AG)的比较:二线研究入组555例他莫昔芬失败患者, 分别每天服来曲唑2.5mg、0.5 mg和AG 250 mg, 3组有效率分别为19.5%、16.7%和12.4%, 有效和稳定的持续时间分别为21个月、18个月和14个月。来曲唑2.5 mg组的肿瘤进展时间(TTP)、治疗失败时间TTF和总生存均明显优于AG组,也略优于0.5 mg组。
新辅助治疗:研究显示他莫昔芬治疗过的患者发生对侧乳腺癌的几率低于对照组, 由此发现他莫昔芬有预防乳腺癌的作用。但只是阻断雌激素的作用, 而不能减少其合成, 所以无法去除雌激素代谢产物的潜在致癌作用, 即引起子宫内膜癌变的副作用, 而第三代芳香化酶抑制剂能够抑制雌激素的合成, 弥补他莫昔芬的不足,理论上预防作用更佳。来曲唑国际协作多中心Ⅲ期临床研究,绝经后、ER(雌激素受体)和(或)PR(孕激素受体)阳性原发乳腺癌患者, 162例每天用来曲唑2.5 mg。175例每天他莫昔芬20 mg均为4个月。结果显示, 每一项研究终点, 来曲唑组均显著优于他莫西芬组, 有更多的患者适于进行BCS, 45%的患者从保留乳房中获益。来曲唑组患者客观缓解率(ORR)显著高于他莫昔芬组(55% vs36% , P
通过对目前众多的大型临床试验进行综合, 证实来曲唑在绝经后乳腺癌治疗的各阶段, 即在晚期乳腺癌的一线治疗、原发性乳腺癌的新辅助治疗以及早期乳腺癌的后续强化治疗中, 均有卓越的临床疗效, 并且副作用较小, 患者依从性高。在早期乳腺癌的辅助治疗中的作用将在目前正在进行的临床试验中得出结论。一些基因水平的研究将更加深入的揭示出来曲唑的临床应用价值、应用特点, 从而更好地指导来曲唑的临床应用, 使更多的患者从中受益。来曲唑对雌激素具有强烈抑制作用的特点, 也为它开拓其他与女性激素水平有关的癌症治疗领域奠定了基础。
2.2.3阿那曲唑(Anastrozole)
阿那曲唑是第3 代芳香化酶抑制剂, 于1995年经美国食品和药品管理局(FDA)批准用于治疗转移性乳腺癌。在中国, 从1997 年9月至1998 年6月进行了进口前的临床验证研究, 单药治疗绝经后晚期乳腺癌患者, 获得25.0 %的有效率,于1999 年经中国国家药品监督管理局(CFDA)批准正式在国内上市。
阿那曲唑适用于经他莫昔芬及其它抗雌激素疗法仍不能控制的绝经后妇女的晚期乳腺癌。对雌激素受体阴性的病人,若其对他莫昔芬呈现阳性的临床反应,可使用阿那曲唑。其可抑制绝经期后患者肾上腺中生成的雄烯二酮转化为雌酮,从而明显地降低血浆雌激素水平,产生抑制乳腺肿瘤生长的作用。另外,阿那曲唑对肾上腺皮质类固醇或醛固酮的生成没有明显影响。
阿那曲唑用于二线解救治疗:大规模临床研究结果显示, 阿那曲唑作为一线药物, 可代替他莫昔芬用于治疗绝经后妇女的晚期乳腺癌, 并可考虑为首选一线治疗。阿那曲唑具有强效芳香化酶抑制作用, 在外周血循环和肿瘤组织内部均能达到近完全雌激素抑制。1998 年完成的一项多中心Ⅲ期临床试验中, 比较阿那曲唑与甲地孕酮治疗既往他莫昔芬失败患者。两种药物取得相似的临床获益率, 阿那曲唑1mg组的中位死亡时间为26 .7 个月, 甲地孕酮组为22 .5 个月。阿那曲唑治疗组在生存率方面明显优于甲地孕酮组。研究结果也显示阿那曲唑治疗晚期乳腺癌的耐受性比甲地孕酮更好。因此阿那曲唑作为二线解救治疗可以取代甲地孕酮。
阿那曲唑用于辅助治疗:阿那曲唑在一线治疗中的数据显示了阿那曲唑在早期乳腺癌治疗中的地位。因此又有阿那曲唑、他莫昔芬单独和联合应用试验(Anast rozole Tamox ifenAlone or Combination , ATAC 试验)研究, 目的通过辅助治疗中与他莫昔芬比较以证实其在早期乳腺癌患者中具有同样的疗效。
阿那曲唑与其他第3 代芳香化酶抑制剂在解救治疗中的对比研究:目前有代表性的第3 代芳香化酶抑制剂主要是甾体类的依西美坦和非甾体类的阿那曲唑、来曲唑。在19个国家、110个中心进行了一项大型研究, 比较来曲唑和阿那曲唑两药用于二线治疗绝经后晚期乳腺癌。研究共有713例患者入组。来曲唑组356 例, 每日口服来曲唑2.5mg ;阿那曲唑组357例,每日口服阿那曲唑1mg。两组病例均为激素受体阳性或受体不明者。研究中每3个月评价疗效。研究结果显示, 来曲唑组的总反应率(OR)较阿那曲唑组高,分别为19 .1 %和12 .3 %(P =0 .014)。来曲唑的临床获益率数值较阿那曲唑组高, 分别为27 %和23 %, 但无统计学差异(P =0 .218)。而两药的肿瘤进展时间TTP、治疗失败时(TTF)、反应持续时间和临床获益时间上无差别。研究显示两药的耐受性良好。
阿那曲唑单药显示出良好的疗效及耐受性, 它已经在乳腺癌的一线、二线治疗,辅助治疗、新辅助治疗中全面超越传统药物他莫昔芬,成为可能替代他莫昔芬的理想药物。
3.中国指南关于芳香酶抑制剂的使用3.1对于没有接受过抗雌激素治疗或无复发时间较长的绝经后复发患者,他莫昔芬、芳香化酶抑制剂或氟维司群都是合理的选择。绝经后复发转移性乳腺癌,一线内分泌治疗的首选为第3代芳香化酶抑制剂,包括阿那曲唑、来曲唑、依西美坦,因为在他莫昔芬治疗失败的复发转移性乳腺癌的二线治疗中,第3代芳香化酶抑制剂比甲地孕酮更有效。在复发转移性乳腺癌的一线内分泌治疗中,第三代的芳香化酶抑制剂明显优于他莫昔芬。绝经前复发转移性乳腺癌患者首选化疗,如果激素受体阳性患者适合或需要用芳香化酶抑制剂进行内分泌治疗时,首选双侧卵巢切除手术,后续联合芳香化酶抑制剂。药物性卵巢功能抑制联合芳香化酶抑制剂也是可以考虑的方案(但尚缺乏临床证据)。
3.2绝经后的患者,一线内分泌治疗可以选择芳香化酶抑制、氟维司群、他莫昔芬或托瑞米芬。通常会优先选择芳香化酶抑制剂,存在芳香化酶抑制剂治疗禁忌证、曾行芳香化酶抑制剂辅助内分泌治疗且无病生存时间短、或因经济原因不能接受芳香化酶抑制剂治疗的患者,可考虑给予他莫昔芬或托瑞米芬。
3.3可以换用另一类芳香化酶抑制剂。如非甾体类芳香化酶抑制剂(来曲唑、阿那曲唑)治疗失败后,可以考虑换为甾体类芳香化酶抑制依西美坦治疗,反之亦然。2
 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国