

导读
近日,柏林工业大学Martin Oestreich课题组报道了一种铜催化前手性三氟甲磺酸吡啶鎓(pyridinium triflates)与硅亲核试剂(由Si-B试剂中释放生成)的C4-选择性加成反应。其中,使用Cu(CH3CN)4PF6作为预催化剂,(R,R)-Ph-BPE (1,2-bis[(2R,5R)-2,5-diphenylphospholan-1-yl]ethane)作为手性配体,可以优异的对映选择性实现吡啶鎓的去芳构化。同时,C3-位上需存在一个羰基,其可能作为一个弱的基团用于预组织(preorganize)和导向C4-位的亲核进攻。此外,合成的4-硅基化的1,4-二氢吡啶可以进一步转化为官能团化的哌啶衍生物。文章链接DOI:10.1002/anie.202407056.
(图片来源:Angew. Chem. Int. Ed.)
正文
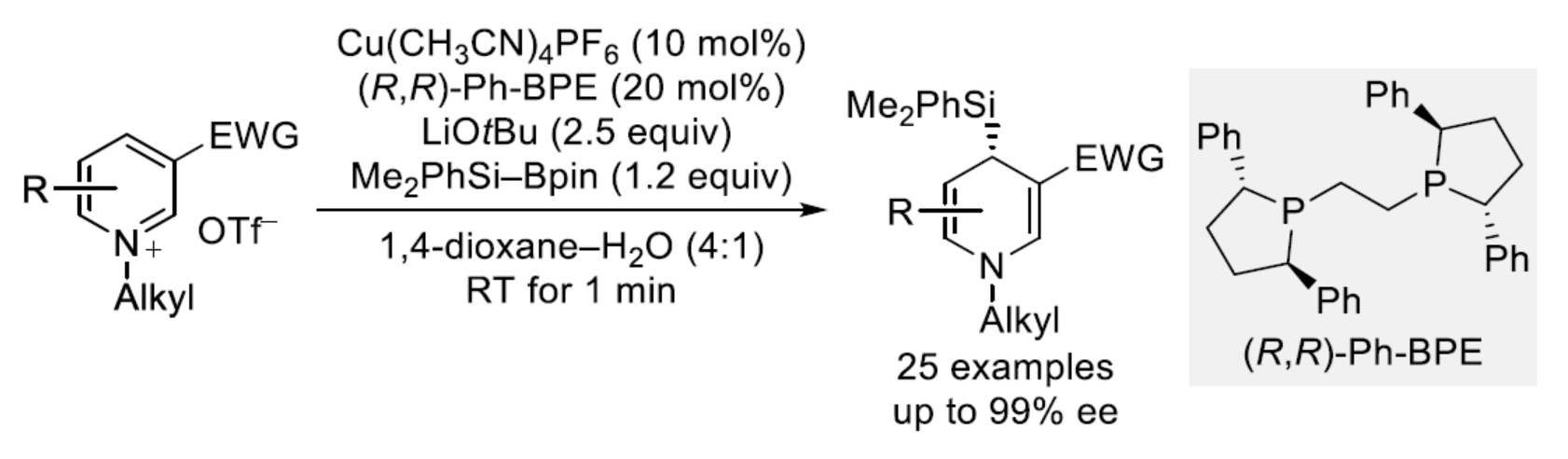
近年来,去芳构化领域备受关注。同时,二维分子通常可转化为富含sp3的三维骨架,具有高度的区域和对映控制性。其中,一个有吸引力的靶点是吡啶核心,因为吡啶的去芳构化可生成药物相关的二氢吡啶,并且在进一步官能团化后,可合成具有价值的哌啶砌块。作为当时具有挑战性的吡啶Birch还原的替代策略,半个世纪前,化学家们已开发了一种还原性1,4-双硅基化反应。后来化学家们意识到,在去硅基化再芳构化之前,所得到的1,4-双硅基化的1,4-二氢吡啶可以用额外的取代基修饰,从而通过临时去芳构化实现吡啶的区域选择性形式亲电取代。鉴于吡啶和吡啶鎓离子的缺电子性质,添加亲核试剂是破坏其芳香性的另一种可能性。早期,利用这一策略,Comins课题组(J. Am. Chem. Soc. 1992, 114, 10972.)完成了在氮原子上带有手性助剂的酰基吡啶鎓离子的C2-选择性硅基化反应。随后,Mangeney课题组(Tetrahedron 1998, 54, 10349.)和Comins课题组(Org. Lett. 2005, 7, 5059.)实现了铜基硅亲核试剂与C3-位具有手性基团的酰基吡啶鎓离子的非对映与C4-选择性加成反应(Scheme 1,top)。然而,对于此类的催化不对称版本仍然缺失。长期以来,Oestreich课题组一直致力于铜催化硅基化反应的研究,该反应使用Si-B试剂作为硅前亲核试剂。Ohmura和Suginome课题组还报道了一种依赖于Pd(0)/Pd(II)催化的不同方法(Scheme 1,middle)。此外,化学家们最近还报道了几种碳亲核试剂参与相关对映和C4-区域选择性加成的例子。基于上述的总结,近日,柏林工业大学Martin Oestreich课题组报道了一种铜催化前手性吡啶鎓离子与硅亲核试剂的C4-选择性去芳构化反应(Scheme 1,bottom)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
(图片来源:Angew. Chem. Int. Ed.)
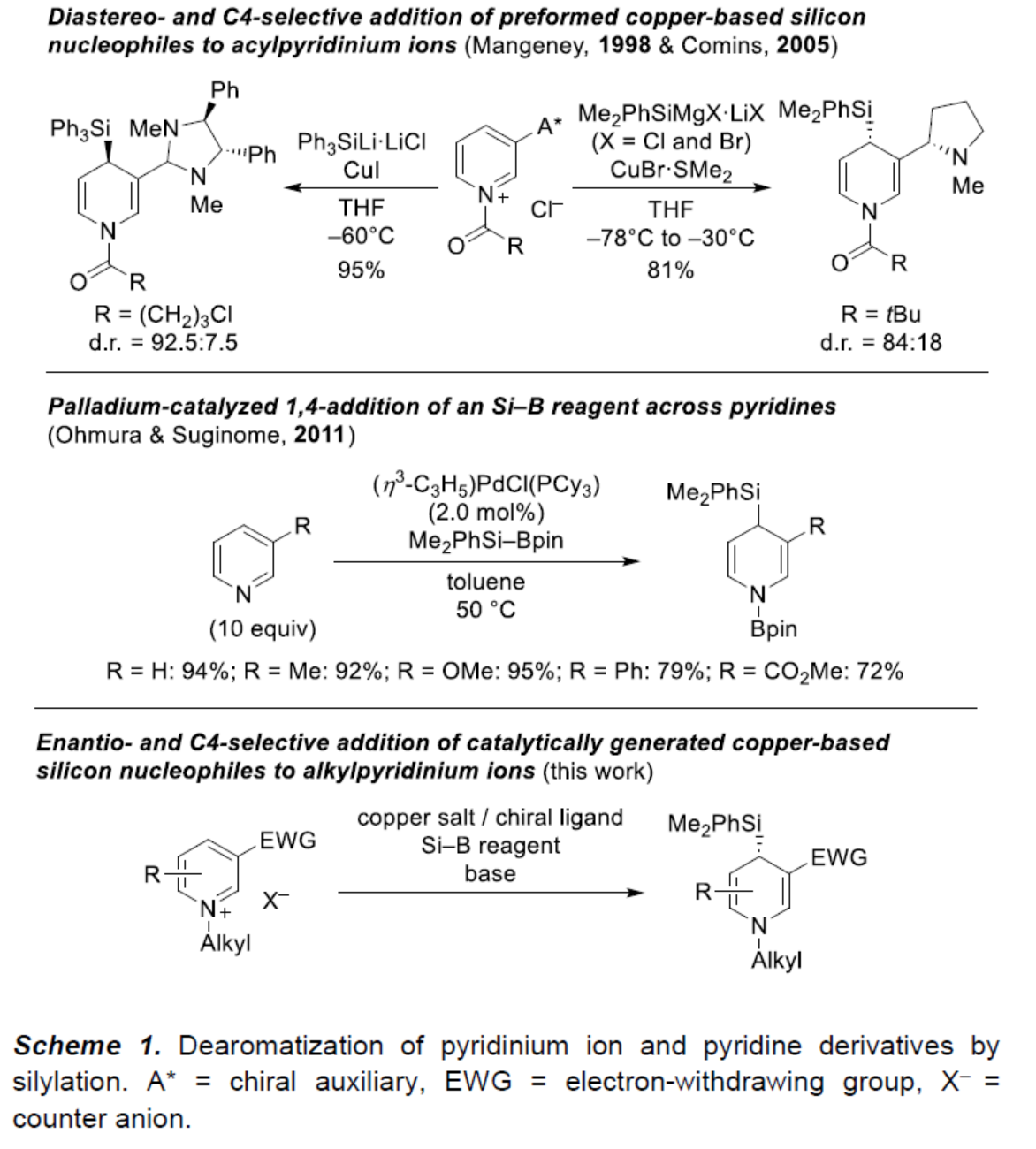
首先,作者以N-甲基吡啶鎓三氟甲磺盐(N-methylpyridinium triflate)1a与Si–B试剂2a作为模型底物,进行了相关去芳构化反应条件的筛选(Table 1)。当底物中的离去基团为OTf,以Cu(CH3CN)4PF6(10 mol %)作为预催化剂,(R,R)-Ph-BPE(20 mol %)作为配体,LiOtBu(2.5 equiv)作为碱,在1,4-dioxane/水(4:1)混合溶剂中室温反应,可以75%的收率得到产物3aa,ee为96%。
(图片来源:Angew. Chem. Int. Ed.)
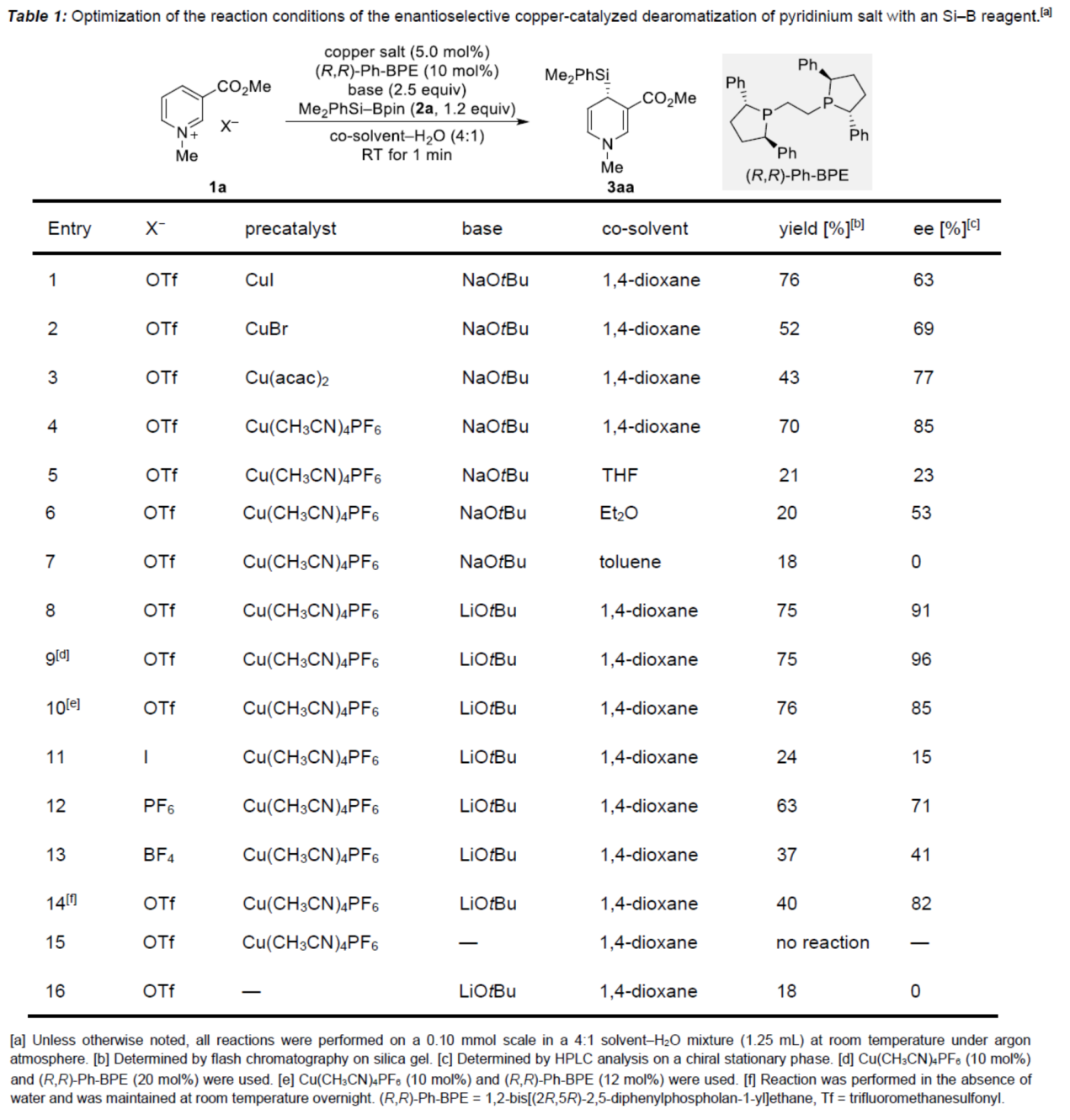
在获得上述最佳反应条件后,作者对其它的3-取代的三氟甲磺酸吡啶鎓进行了研究(Figure 1)。研究结果表明,当底物中的C3-位含有-COMe(4a)时,可以48%的收率和90% ee得到产物5aa。相反,当底物中的C3-位含有-CONH2(6a)与-CONPh2(8a)时,反应的效率较差。同时,当底物中的C3-位含有-CN、-F和-NO2时,反应未能有效的进行。
(图片来源:Angew. Chem. Int. Ed.)
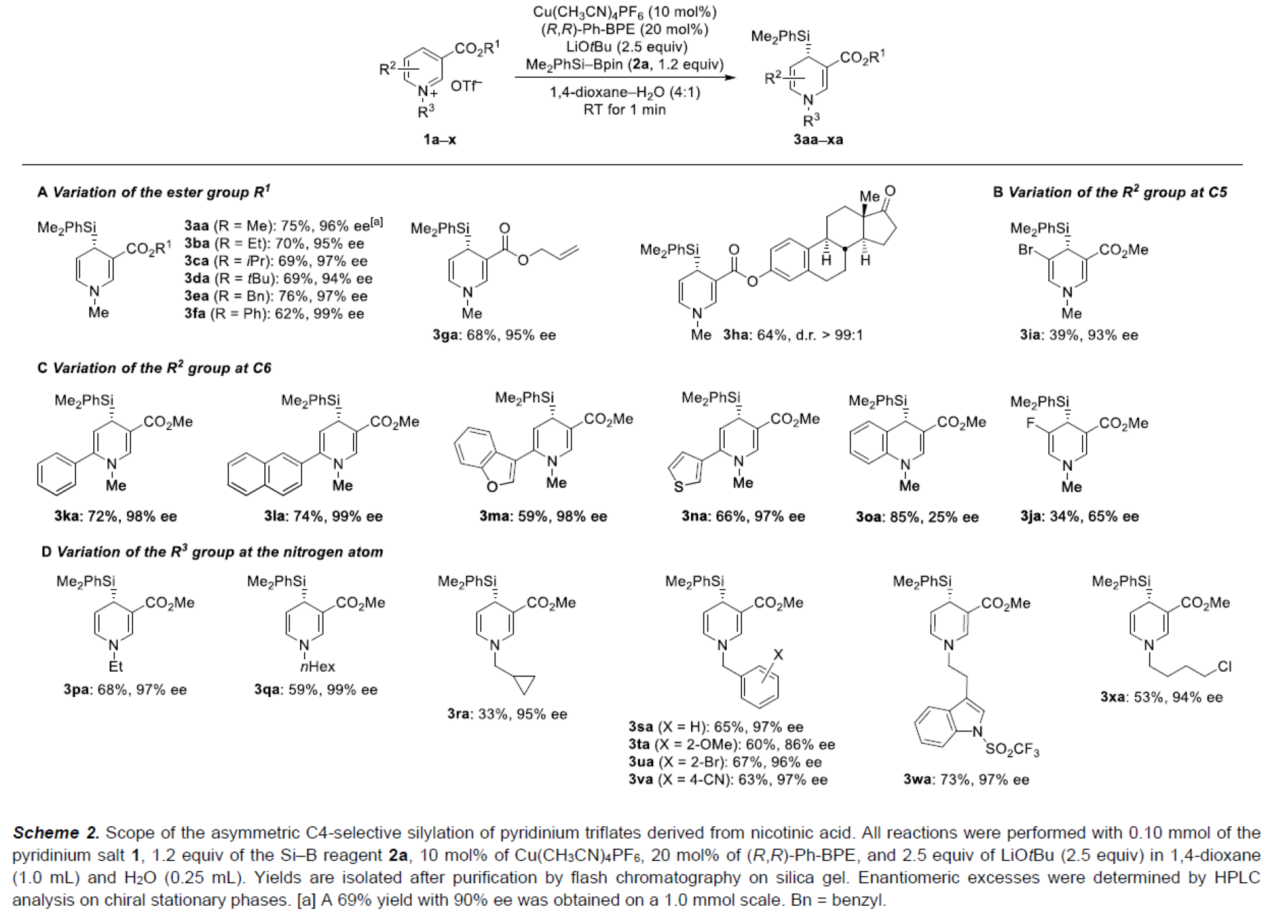
其次,作者对烟酸衍生物的底物的范围进行了扩展(Scheme 3)。首先,当底物1中的R1为各种烷基、芳基、苄基与烯丙基等时,均可顺利反应,获得相应的产物3aa-3ha,收率为62-76%,ee为94-99%。其次,当底物1中的C5-位含有-Br或-F时,反应的效率有所降低,可以39%的收率得到产物3ia(93%ee)和34%的收率得到产物3ja(65%ee)。当底物1中的C6-位含有各种杂芳基时,也与体系兼容,获得相应的产物3ka-3na,收率为59-74%,ee为97-99%。具有喹啉骨架的底物,也是合适的底物,可以85%的收率得到产物3na,但ee仅为25%。此外,当底物1中的R3为各种烷基与苄基时,均可顺利进行反应,获得相应的产物3pa-3xa,收率为33-73%,ee为86-99%。
(图片来源:Angew. Chem. Int. Ed.)
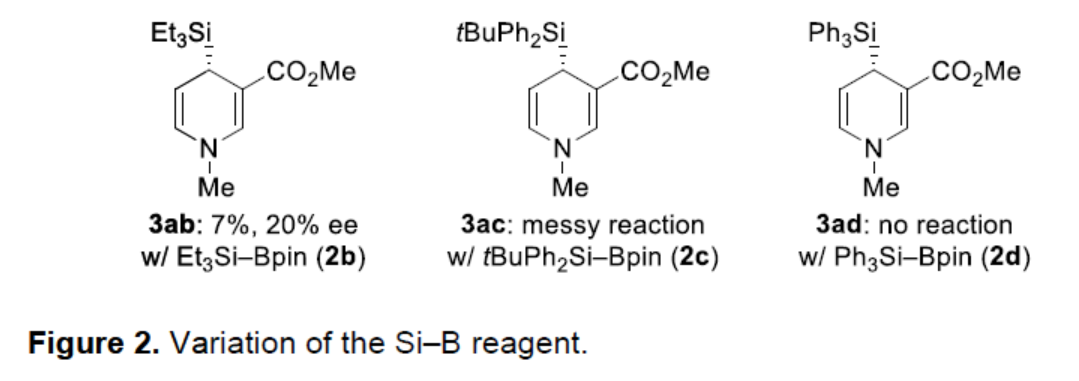
同时,作者还对Si–B试剂的范围进行了考察(Figure 2)。研究表明,Me2PhSi-Bpin(2a)远远优于其他Si-B试剂(2b、2c和2d)。只有Et3Si-Bpin(2b)以低收率得到了1,4-二氢吡啶3ab,但ee值很低。位阻较大的tBuPh2Si-Bpin(2c)得到复杂的反应混合物,而Ph3Si-Bping(2d)根本没有反应。
(图片来源:Angew. Chem. Int. Ed.)
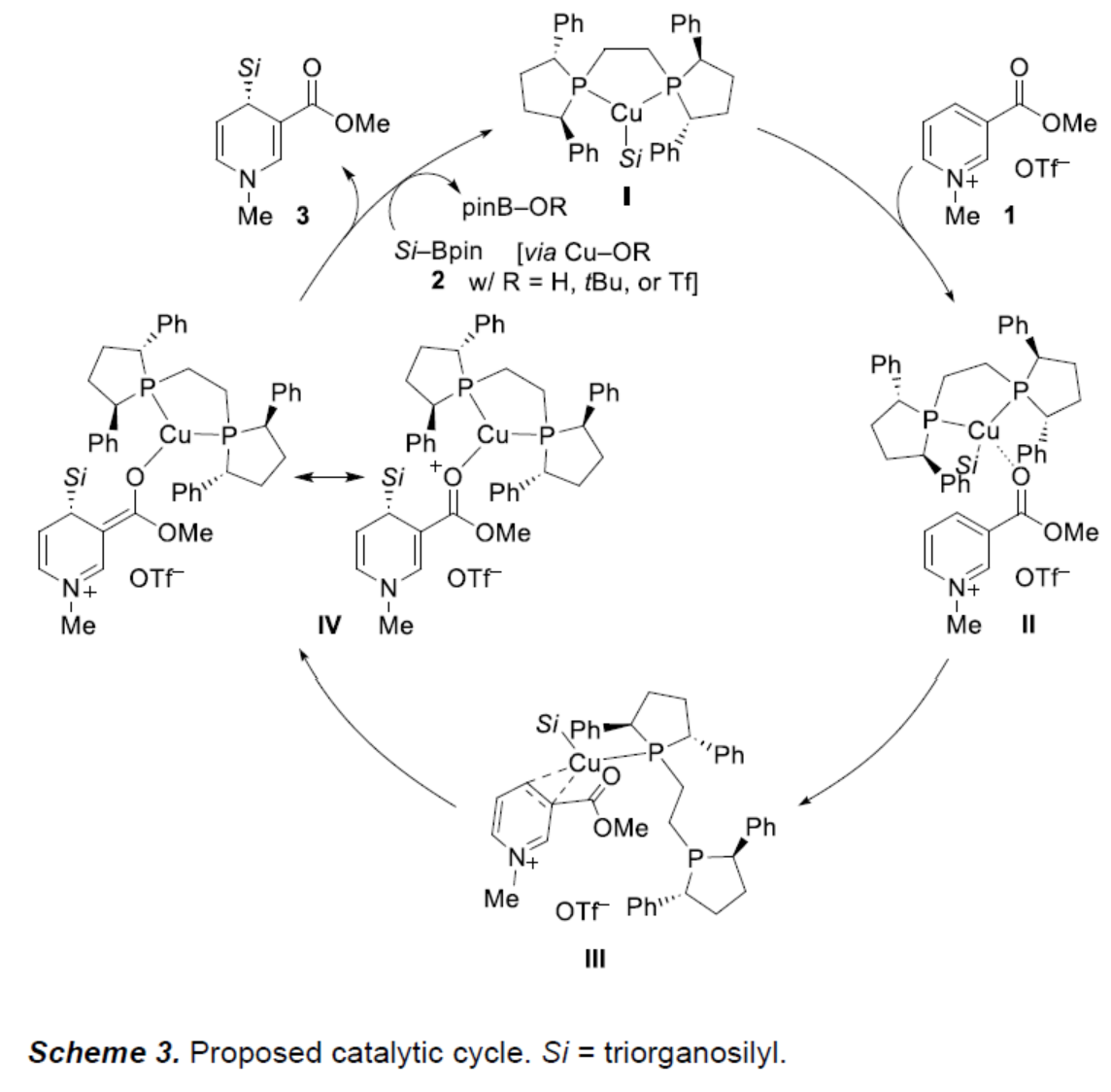
基于相关文献的查阅,作者提出了一种合理的催化循环过程(Scheme 3)。首先,在醇盐碱和水的存在下,铜(I)预催化剂和双齿(R,R)-Ph-BPE通过Si-B试剂2的转金属化生成催化活性的硅基铜(I)配合物I。配合物I中的羰基不仅活化吡啶鎓离子,而且作为导向基团生成弱Lewis对II。同时,Lewis对II与另一种弱加合物III平衡,其中C3和C4之间的吡啶环通过铜(I)中心配位。(R,R)-Ph-BPE配体在预组织(preorganized)III中可以是单齿的,并且磷供体可以帮助将软硅亲核试剂递送到C4-位,生成中间体IV。中间体IV中阳离子(R,R)-Ph-BPE–copper(I)单元解离后,生成目标的二氢吡啶产物3,并再生铜(I)催化剂。该步骤涉及通过氢氧化物(来自水)、醇盐或三氟甲磺酸盐形成的阴离子氧亲核试剂,并且可能在水存在下加速。这些得到的中性铜(I)配合物中的任何一个都与Si-B试剂2进行另一次转金属化催化,以关闭催化循环。
(图片来源:Angew. Chem. Int. Ed.)
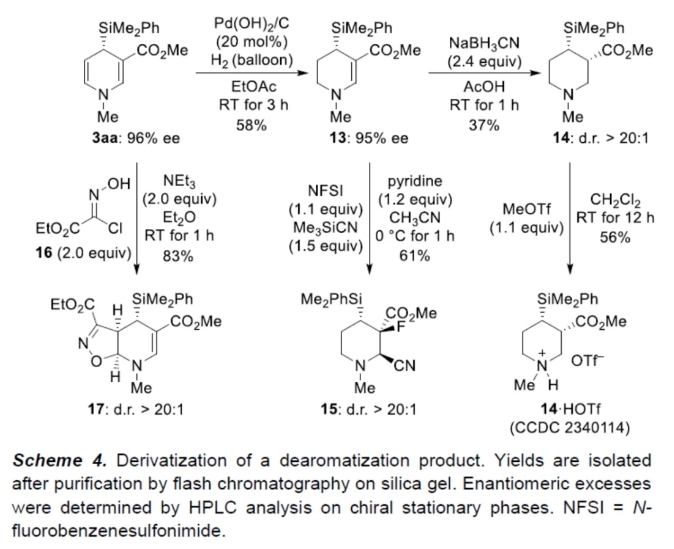
紧接着,作者对反应的实用性进行了研究(Scheme 4)。首先,3aa中的富电子双键可以进行化学选择性氢化,可以58%的收率得到环己烯衍生物13,ee为95%。化合物13通过进一步的还原反应,可以37%的收率得到哌啶衍生物14,dr > 20:1。化合物14与MeOTf进行成盐,可以56%的收率得到铵盐14·HOTf。其次,化合物13、NFSI与Me3SiCN在吡啶/乙腈条件下进行氟氰化反应,可以61%的收率得到哌啶衍生物15,dr > 20:1。此外,化合物3aa也可与肟化合物16进行1,3-偶极环加成反应,可以83%的收率得到杂环化合物17,dr > 20:1。
(图片来源:Angew. Chem. Int. Ed.)
总结
柏林工业大学Martin Oestreich课题组报道了一种高效的不对称合成4-硅基-1,4-二氢吡啶的方法,其对映选择性高达99%。该方法基于Cu-O-介导的Si-B试剂(作为硅前亲核试剂)的转金属化进行的。由此生成的铜基硅亲核试剂以优异的区域选择性与活化的三氟甲磺酸吡啶鎓进行加成。C3-位羰基导向的硅亲核试剂在C4-位进行加成。合成的4-硅基化的1,4-二氢吡啶是有价值的合成中间体。
文献详情:
Enantioselective Dearomatization of Pyridinium Salts by Copper-Catalyzed C4-Selective Addition of Silicon Nucleophiles.
Yao Xiao, Zhi-Yuan Zhao, Sebastian Kemper, Elisabeth Irran, Martin Oestreich*.
Angew. Chem. Int. Ed. 2024
https://doi.org/10.1002/anie.202407056




 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国