

导读
近日,美国布兰迪斯大学Chi P. Ting课题组报道了一种抗菌肽(antimicrobial sactipeptide)Enteropeptin A的全合成及其结构确定。Enteropeptin A中含有一个具有未确定立体化学构型的硫代氨基缩酮(thioaminoketa)基团,该基团嵌入一个极不寻常的硫代吗啉(thiomorpholine)环中。在该合成中,涉及通过二硫代磷酸(dithiophosphoric)催化含有脱氢氨基酸和侧半胱氨酸残基的线性肽的MarkovnikovHydrothiolation过程。这种环化反应形成Enteropeptins中的中心硫代吗啉环。在Enteropeptin A未确定的硫代氨基缩酮立体中心制备了两种非对映异构体,并且将它们与真实标准进行比较,并确认了天然产物的立体化学为D构型。Enteropeptin A的全合成代表了迄今为止塞克肽类(sactipeptide)的首次全合成。文章链接DOI:10.1021/jacs.4c06126
(图片来源:J. Am. Chem. Soc.)
正文
塞克肽(sactipeptides)是核糖体合成和翻译后修饰肽(RiPPs)的一个亚类,由硫与α-碳键定义,称为sactionine。许多塞克肽具有抗菌活性,是一类很有前途的新型抗菌化合物。塞克肽是从线性核糖体肽开始生物合成的,该肽在半胱氨酸的硫醇和另一个氨基酸残基的α-碳之间进行环化。所得到的环肽含有立体确定的硫代氨基缩酮,在该天然产物家族的L和D立体化学构型中都发现了这种缩酮。化学加——科学家创业合伙人,欢迎下载化学加APP关注。
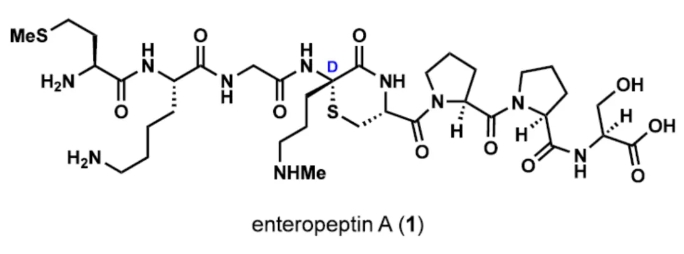
Enteropeptins A−C(1−3)是2022年从人类肠道微生物盲肠肠球菌中分离出来的塞克肽(Figure 1a)。Enteropeptin A(1)是一种窄谱抗菌剂,对其产生的生物体具有抑菌活性。Enteropeptins B(2)和C(3)是其相关的化合物,它们与1的区别分别在于单个氨基酸的缩短或延伸。然而,由于Enteropeptins A的分离产率较低,天然产物尚未通过NMR直接表征,并且在其硫代氨基缩酮碳上具有未确定的立体化学构型。2003年,Vederas课题组(J. Am. Chem. Soc.2003, 125, 4726.)首次合成了线性Sactionine。2022年,Malins课题组(Org. Lett. 2022, 24, 3680.)报道了通过α-溴甘氨酸衍生物与半胱氨酸硫醇的烷基化合成含thioaminal的肽,这是获得塞克肽的一种潜在策略。到目前为止,还没有关于塞克肽天然产物的化学合成的报道。
首先,作者进行了逆合成分析(Figure 1b)。Enteropeptin A可由中心硫代吗啉砌块5与N-末端二肽砌块4和C-末端三肽砌块6通过酰胺键形成反应构建。砌块5可由砌块7经分子内的环化反应制备。砌块7可由脱氢氨基酸9经烯酰胺的质子化制备,涉及使用Brønsted酸催化剂(DTPA)。

(图片来源:J. Am. Chem. Soc.)
epi-Enteropeptin A(epi-1)的全合成(Scheme 1)。以N-苄氧羰基-2-磷酰甘氨酸三甲酯(10)为初始底物,在Pd/C/H2/MeOH条件下进行脱保护,可以几乎定量的收率得到砌块11。砌块11与Phth-Gly-OH(12)在NMM(N-甲基吗啉)/IBCF(氯甲酸异丁酯)/THF条件下进行偶联反应,可以81%的收率得到二肽砌块13。砌块13与醛化合物14在TMG(四甲基胍)/DCM条件下进行Horner−Wadsworth−Emmons反应,可以80%的收率得到砌块15。砌块15在LiI/EtOAc条件下进行去甲基化反应,可以56%的收率得到羧酸砌块16。砌块16与L-Cys(STrt)-OMe(17)在EDCI(1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐)/HOAt/NMM(N-甲基吗啉)条件下进行酰胺形成反应,可以61%的收率得到砌块18。同时,砌块18在TFA/TIS/DCM条件下进行脱保护反应,可以92%的收率得到硫醇砌块19。然而,利用砌块19制备砌块20的多种策略,均未能成功。通过对反应条件的大量尝试后发现,当以二硫代磷酸8(0.05 equiv)作为催化剂,PhCF3作为溶剂,在微波条件下150 oC反应2 h,可以60%的收率得到砌块20,是单一的非对映异构体(Table 1)。值得注意的是,砌块20的X-射线晶体分析表明,该过程发生了环化反应,仅得到L-硫代氨基缩酮。砌块20在H2NCH2CH2NH2/CH2Cl2/MeOH条件下选择性的去除邻苯二甲酰亚胺保护基,并在NMM/HOAt/EDC·HCl条件下进行偶联反应,可以两步61%的总收率得到五肽砌块21。砌块21在LiOH/MeOH/H2O条件下进行甲酯的水解,并与胺化合物6在EDC·HCl/HOAt/NMM/DMF条件下进行酰胺形成反应,可以两步71%的总收率得到八肽砌块S3。砌块S3在TMSBr/茴香硫醚/TFA条件下进行全脱保护反应,可以59%的收率得到epi-1。
Enteropeptin A(1)的全合成(Scheme 1)。利用ent-17制备的ent-20为底物,其含有D-硫代氨基缩酮以及在半胱氨酸α-立体中心具有D-立体化学构型。ent-20在DBU/DCM条件下进行三次循环的差向异构化,可以72%的总收率得到砌块23。砌块23在LiI/EtOAc条件下去甲基化反应,并与胺化合物6在EDC·HCl/HOAt/NMM/DMF条件下进行偶联反应,可以两步76%的总收率得到六肽砌块25。砌块25在H2NCH2CH2NH2/CH2Cl2/MeOH条件下选择性的去除邻苯二甲酰亚胺保护基,并与化合物4在NMM/HOAt/EDC·HCl条件进行偶联反应,可以两步53%的总收率得到砌块26。砌块26在在TMSBr/茴香硫醚/TFA条件下进行全脱保护反应,可以81%的收率得到Enteropeptin A(1)。
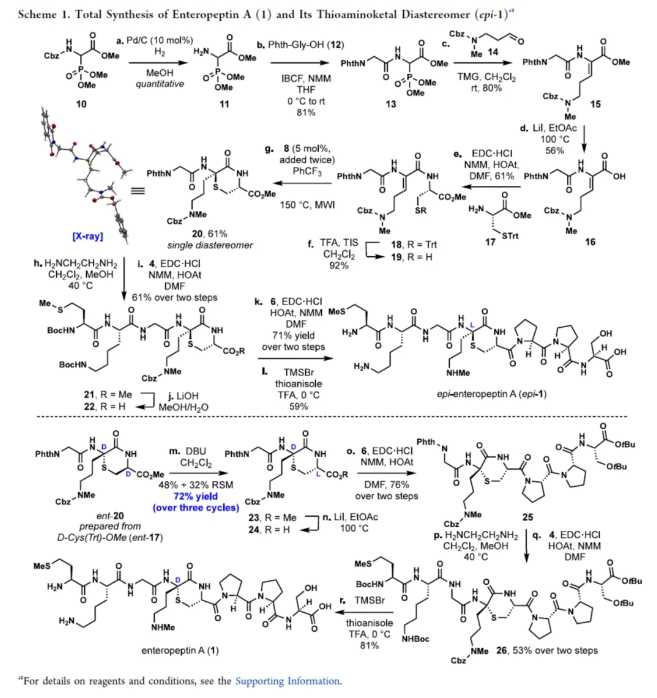
(图片来源:J. Am. Chem. Soc.)
砌块19制备砌块20的Markovnikov Hydrothiolation条件筛选(Table 1)。
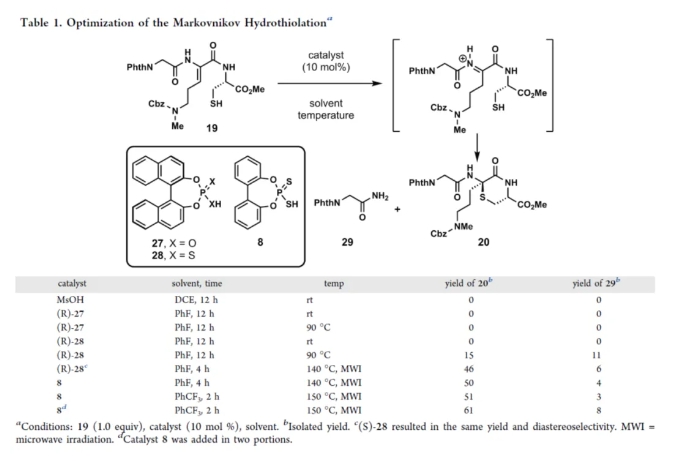
(图片来源:J. Am. Chem. Soc.)
在获得上述1和epi-1后,作者对Enteropeptin A的结构进行了结构鉴定(Figure 2)。通过真实与合成的Enteropeptin A的LC/MS分析结果表明,真实的Enteropeptin A与合成的1(D-硫代氨基缩酮)完全重叠,但与合成的epi-1(L-硫代氨基缩酮)完全不重叠。这些结果表明,Enteropeptin A具有D-硫代氨基缩酮单元。
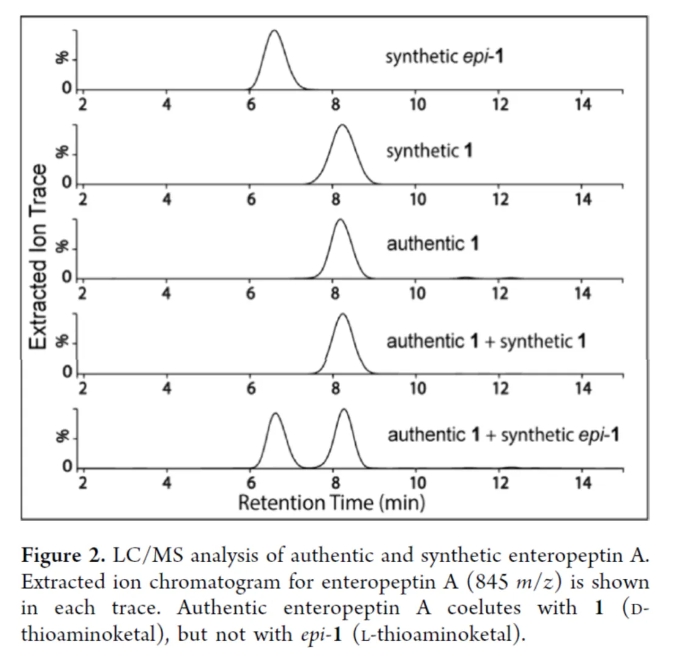
(图片来源:J. Am. Chem. Soc.)
总结
美国布兰迪斯大学Chi P. Ting课题组报道了一种14步全合成Enteropeptin A的路线。其中,有机催化非对映选择性环化以形成硫代吗啉环骨架是反应的关键。随后,半胱氨酸α-质子的差向异构化产生用于合成Enteropeptin A的所需非对映异构体。鉴于肽治疗领域的不断扩大,立体选择性产生环肽的方法将极大地促进其制备,并助力评估其生物活性。除了RiPPs,作者还报道了非核糖体肽以及含有硫代氨基缩酮的epidithiodiketopiperazines(ETPs)的立体选择性肽合成的其他例子。本文的合成描述了基于enteropeptin类天然产物合成硫代吗啉肽的模块化方法。随着新一类RiPPs的发现,从复杂分子合成的角度来看,开发合成策略以获得这一快速增长的生物活性天然产物家族将是非常有可能。
文献详情:
Yiwei Zhang, Shuvendu Saha, Yannik C. C. Esser, Chi P. Ting*. Total Synthesis and Stereochemical Assignment of Enteropeptin A. J. Am. Chem. Soc.2024, https://doi.org/10.1021/jacs.4c06126

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国