

导读
近日,加拿大皇后大学(Queen’s University)Cathleen M. Crudden课题组与日本名古屋大学(Nagoya University)Masakazu Nambo课题组联合发展了镍催化α-芳基环砜与格氏试剂的对映特异性交叉偶联反应,实现了对映体富集开环芳基化。尽管格氏试剂的碱性很强,但作者仍可以观察到高达99%的手性转移。原位监测实验表明,交叉偶联与去质子化在动力学上是竞争的,因此可以实现高对映选择性转化。此结果是对现有文献的重要补充,因为其展示了砜的对映特异性交叉偶联以及当交叉偶联反应以足够快的速度发生时通过去质子化来规避消旋化过程的能力。相关成果发表在Nat. Chem.上,文章链接DOI:10.1038/s41557-024-01594-x。

(图片来源:Nat. Chem.)
正文
发展可以有效控制立体化学的碳-碳键形成方法对构建复杂分子至关重要。而交叉偶联反应是构建分子最有效和最广泛使用的反应之一,这些反应可以保持手性或引入手性,是合成工具箱中的强大的“工具”。砜是一种简单易得的有机亲电试剂,其具有许多作为交叉偶联反应中偶联配偶体的特征。然而,自1979年化学家首次使用它们以来,还没有在对映选择性、对映特异性或对映收敛的交叉偶联中使用它们的例子。近日,加拿大皇后大学(Queen’s University)Cathleen M. Crudden课题组与日本名古屋大学(Nagoya University)Masakazu Nambo课题组成功实现了环砜与格氏试剂的对映特异性交叉偶联反应(Fig. 1)。化学加——科学家创业合伙人,欢迎下载化学加APP关注。

(图片来源:Nat. Chem.)
首先,作者以连有萘基取代的砜rac-1a作为模板底物对反应进行探索(Table 1)。通过一系列条件筛选,作者发现当使用rac-1a (0.1 mmol), PhMgBr (2) (2.5 equiv.), [NiBr2(DMPMA)]2 (5 mol%), 在THF (0.3 M)中75 °C反应16 h, 随后加入 MeI (10 equiv.),在DMSO (0.2 M)中85 °C反应4 h,可以以83%的分离产率得到产物3aa(entry 9)。控制实验表明,在没有镍催化剂和配体存在下反应是不发生的(entry 10, 11)。

(图片来源:Nat. Chem.)
在得到了最优反应条件后,作者对此转化的底物兼容性进行了考察(Table 2)。实验结果表明一系列不同取代的芳基格氏试剂(Table 2左边)和砜(Table 2右边)均具有良好的兼容性,以16-83%的产率得到相应的产物3aa-3ha。此外,作者还对不同环尺寸的砜进行了考察,发现四元环(1i)和六元环砜(1j)同样是有效的偶联配偶体,但产率相对较低(3ia, 34%; 3ja, 32%)。有趣的是,四倍过量的格氏试剂的使用对六元环砜的开环是最有效的,这需要抑制β-氢消除。此现象表明亚磺酸盐在开环后仍与金属结合,并影响交叉偶联的倾向,而不是β-氢消除。
接下来作者探讨了亚磺酸盐捕获试剂的兼容性(Table 2底部)。首先,作者以1 g规模成功制备了萘类似物5aa,产率86%。这些反应在一锅中进行,不需要分离或纯化中间体。亚磺酸盐的分离是极其重要的,因为它可以与各种亲电试剂反应,来制备取代的砜和磺酰胺。随后,作者考察了交叉偶联产物与其它亲电试剂的反应,包括与烷基溴和烯丙基溴的直接反应,分别以73%和67%的产率得到二烷基砜产物6aa和7aa。在缺电子溴吡啶上的SNAr反应也受到影响,以中等产率(32%)得到杂芳基砜8aa。与SO2Cl2反应可原位生成磺酰氯衍生物,且与吗啡啉反应时,可以47%的产率得到磺酰胺9aa。

(图片来源:Nat. Chem.)
在证明了开环交叉偶联反应的有效性后,作者接下来将目光转向其对映体富集类似物。作者采用Corey-Bakshi-Shibata还原法得到二醇11,并进行了甲磺酰化和Na2S处理,随后通过氧化得到对映体富集的砜(S)-1a,且产物结构得到了单晶X-射线衍射分析的确证(Fig. 2a)。实验结果表明,对映体富集的底物(S)-1a与2a的开环芳基化可以以≥99%的对映体特异性进行。
为了确定产物的绝对构型,作者制备了(R)-3aa(Fig. 2b)。通过与镍催化的交叉偶联反应所制备的(R)-3aa比较,作者得出镍催化体系将(S)-1a转化为(S)-3aa是构型反转的。这一观察结果与其它交叉偶联反应的文献报道相一致。不同的有机镁试剂(2b和2f-2h)也同样具有良好的对映体特异性,并且在使用1a的(R)或(S)对映体的实验中获得了类似的结果(Fig. 2c)。对映体富集的二取代环砜14也可成功实现转化,得到相应的产物ent-15,且具有良好的对映特异性和非对映特异性。
接下来,作者通过1H NMR和手性超临界流体色谱(SFC)分析,探索了反应速率以及原料和产物的对映纯度随时间的变化(Fig. 2d,e)。由于在75 oC时反应速度太快而无法监测,因此作者将反应温度降至30 oC。在此条件下,在反应的前5分钟内,(S)-3aa的转化率达到(S)-1a消耗的90%以上,立体化学保留率 > 99%(Fig. 2d)。底物(R)-3ag需要> 45分钟才能达到类似的阶段,其对映体特异性较低(79%; Fig. 2e)。

(图片来源:Nat. Chem.)
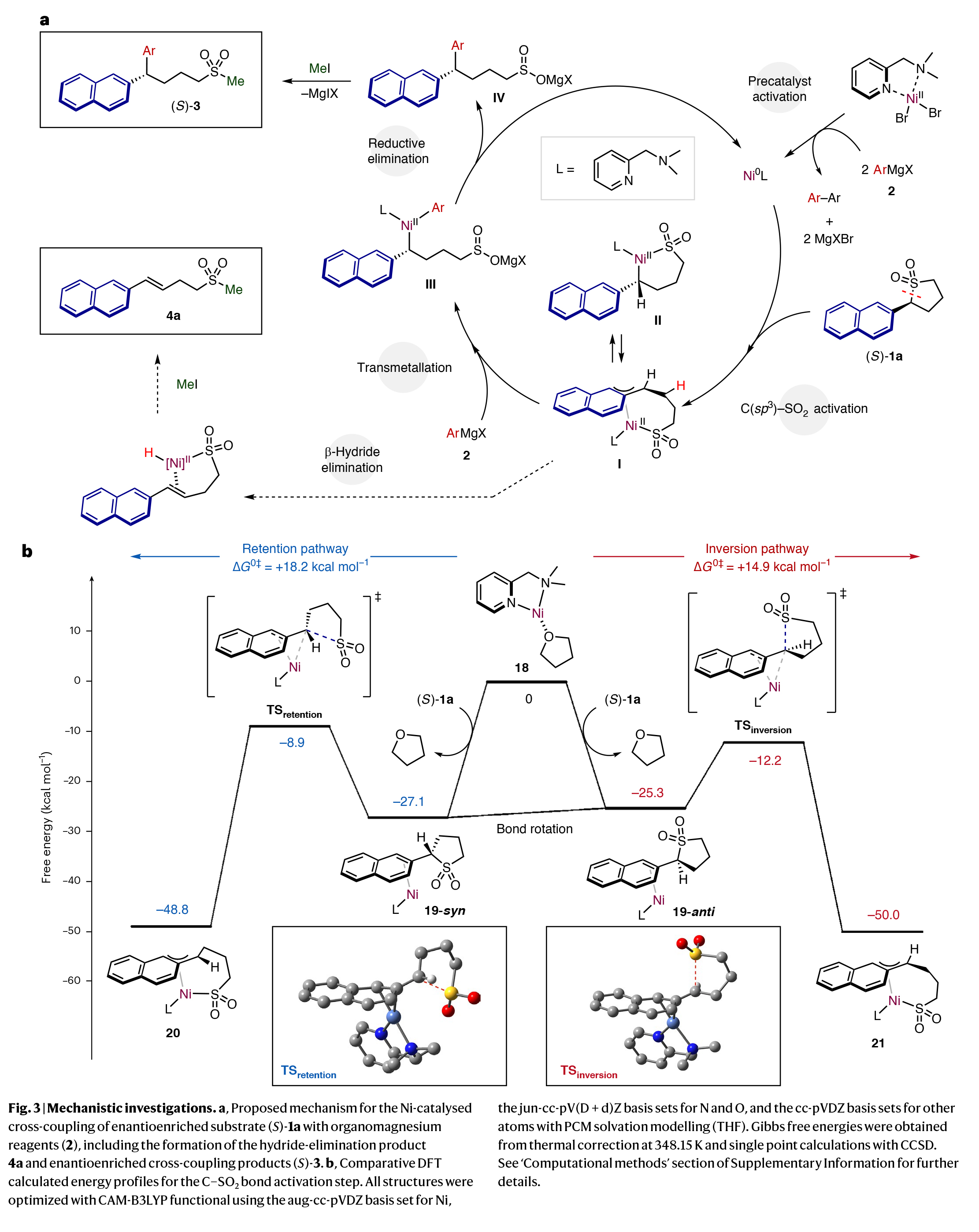
基于上述观察结果,作者提出了此转化可能的反应机理(Fig. 3a):催化循环由镍预催化剂[NiBr2(DMPMA)]2与两当量的格氏试剂引发,在消除相应的自偶联(联芳基)产物后,得到具有催化活性的Ni0(DMPMA)物种。接下来,α-萘基取代的环砜(S)-1a在扩展的π-芳香体系的促进下,在最活泼的C(sp3)-SO2键上进行氧化加成。反应的立体特异性可能是由氧化加成的性质控制的,因为转金属化和还原消除通常伴随着构型的保持。
一种潜在的氧化加成反应机理涉及亲核镍络合物与砜通过直接的SN2反应而不涉及π-体系。基于此转化严格依赖于底物中π扩展芳烃的存在,并考虑到所采用的底物的位阻,这种机理被认为是不可能的。
相反,催化剂很可能与底物络合,然后发生顺式或反式的氧化加成,生成π-萘基镍物种。这两种可能性在文献中均有报道。在期望的反应途径中,氧化加成(I/II)的产物随后与格氏试剂发生反应,预计这将在立体化学保留的情况下发生,并再生活性Ni0物种。最后,IV与MeI反应生成芳基化产物(S)-3aa。或者,中间体I/II可以在甲基化后进行β-氢消除得到4a。
(图片来源:Nat. Chem.)
总结
Cathleen M. Crudden课题组与Masakazu Nambo课题组以芳基格氏试剂为亲核试剂,发展了镍催化α-芳基环砜的对映体富集开环芳基化反应。重要的是环砜可以将磺酰基保留在产物中,因为砜和磺酰胺骨架是普遍存在的官能团。根据格氏试剂的性质,此交叉偶联反应可以以高达99%的对映体特异性实现转化。此外,作者探讨了相对于去质子化来说,交叉偶联反应速度的重要性。
文献详情:
Enantiospecific cross-coupling of cyclic alkyl sulfones.
Roberto Nolla-Saltiel, Zachary T. Ariki, Stefanie Schiele, Jana Alpin, Yasuyo Tahara, Daisuke Yokogawa, Masakazu Nambo,* Cathleen M. Crudden*.
Nat. Chem., 2024
https://doi.org/10.1038/s41557-024-01594-x.

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国