

导读
最近,化学家们发现降西松烷类(norcembranoids)化合物Scabrolides A和B具有明显不同的碳三环骨架。因此,合成Scabrolide A的路线不能扩展到其衍生化天然产物的合成。近日,德国马克斯普朗克煤炭研究所Alois Fürstner课题组设计了一种新型的方法,该方法依赖于酮的分子内烯基化,在桥头烯酮位点形成空间拥堵的中心环庚烯环。通过镧系元素催化的Mukaiyama-Michael加成反应构建所需的环化前体。然后,通过烯酮的烯丙基重排/氧化来引入不协调(dissonant)的1,4-氧合模式,然后进行同面(suprafacial)上1,3-转位反应。合成的Scabrolide B通过脱水转化为Sinuscalide C,并通过碱介导的异构化/oxa-Michael加成转化为Ineleganolide,这具有潜在的生物合成意义。在碱性条件下,后者通过复杂的仿生串联反应可转化为Horiolide。文章链接DOI:10.1021/jacs.4c09467
(图片来源:J. Am. Chem. Soc.)
正文
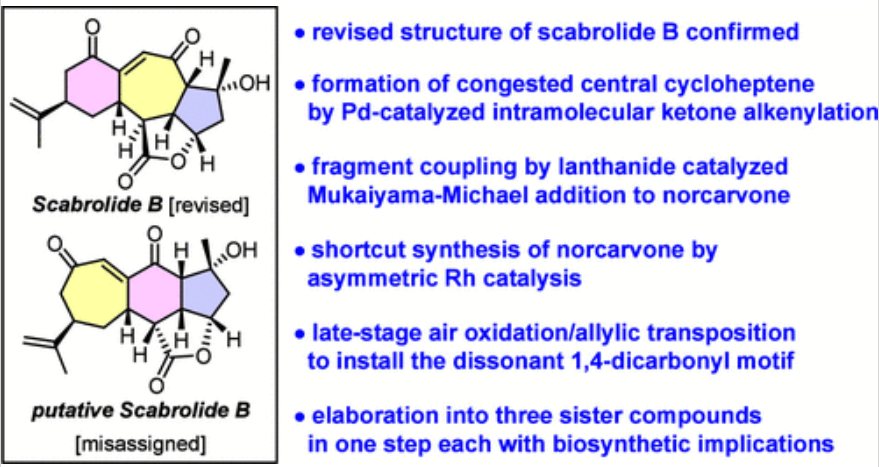
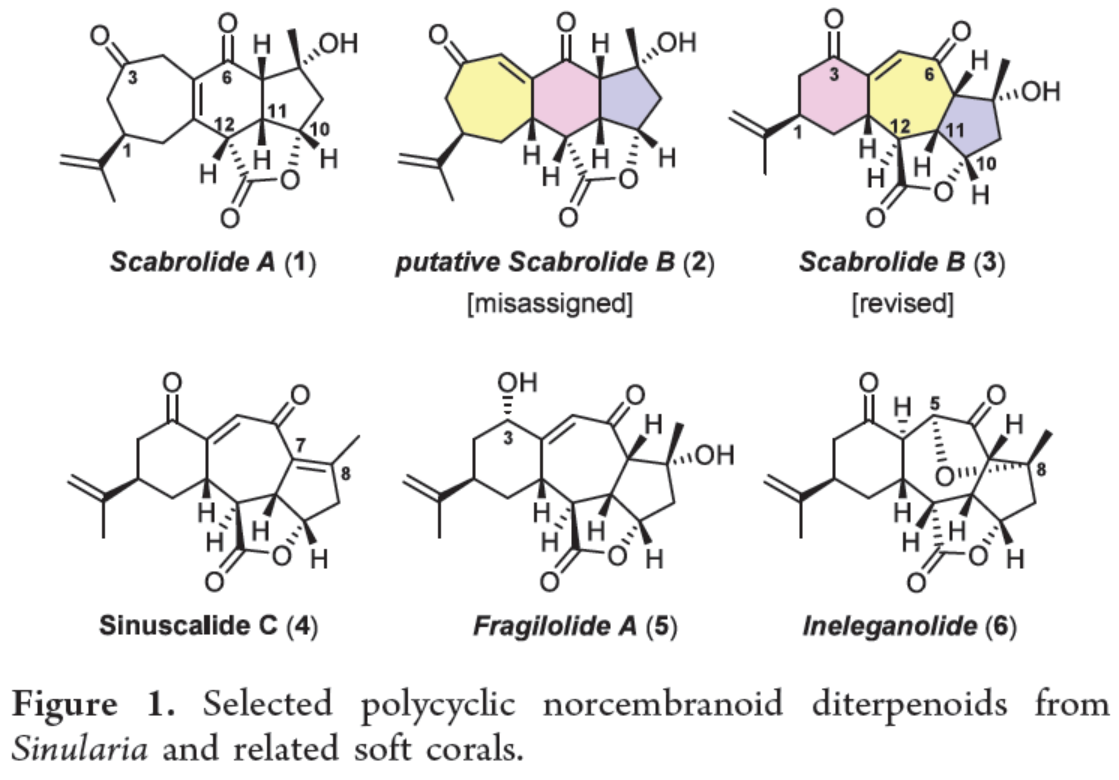
Scabrolide A(1)是一种有趣的降西松烷类化合物,来源于Sinularia属的软珊瑚。利用了早期的生物合成考虑,1是通过双键异构化从Scabrolide B(2)中衍生出来的(Figure 1)。虽然这种转化可以几乎定量的收率实现,但作者注意到了一个令人困惑的不一致:合成1与Scarrolide A完全对应,但其前体2与推测的Scarrolid B根本不匹配。基于DFT计算研究发现,作者对Scarrolid B(2)的结构进行了修订,重新定义为Scarrolid B(3)。天然产物Scabrolide B(3)与Scabrolide A(1)的显著不同之处在于,它具有6−7−5而不是7−6−5的碳三环骨架。因此,它与Sinuscalide C(4)及其脱水子类密切相关,也与Fragilolide A(5)密切相关,其中C3酮被还原。除了这种结构差异之外,1和3的C12立体中心具有相反的构型,从而能够将Scabrolide B(3)与Inelenolide(6)区分开来。化学加_合成化学产业资源聚合服务平台,欢迎下载化学加APP关注。
(图片来源:J. Am. Chem. Soc.)
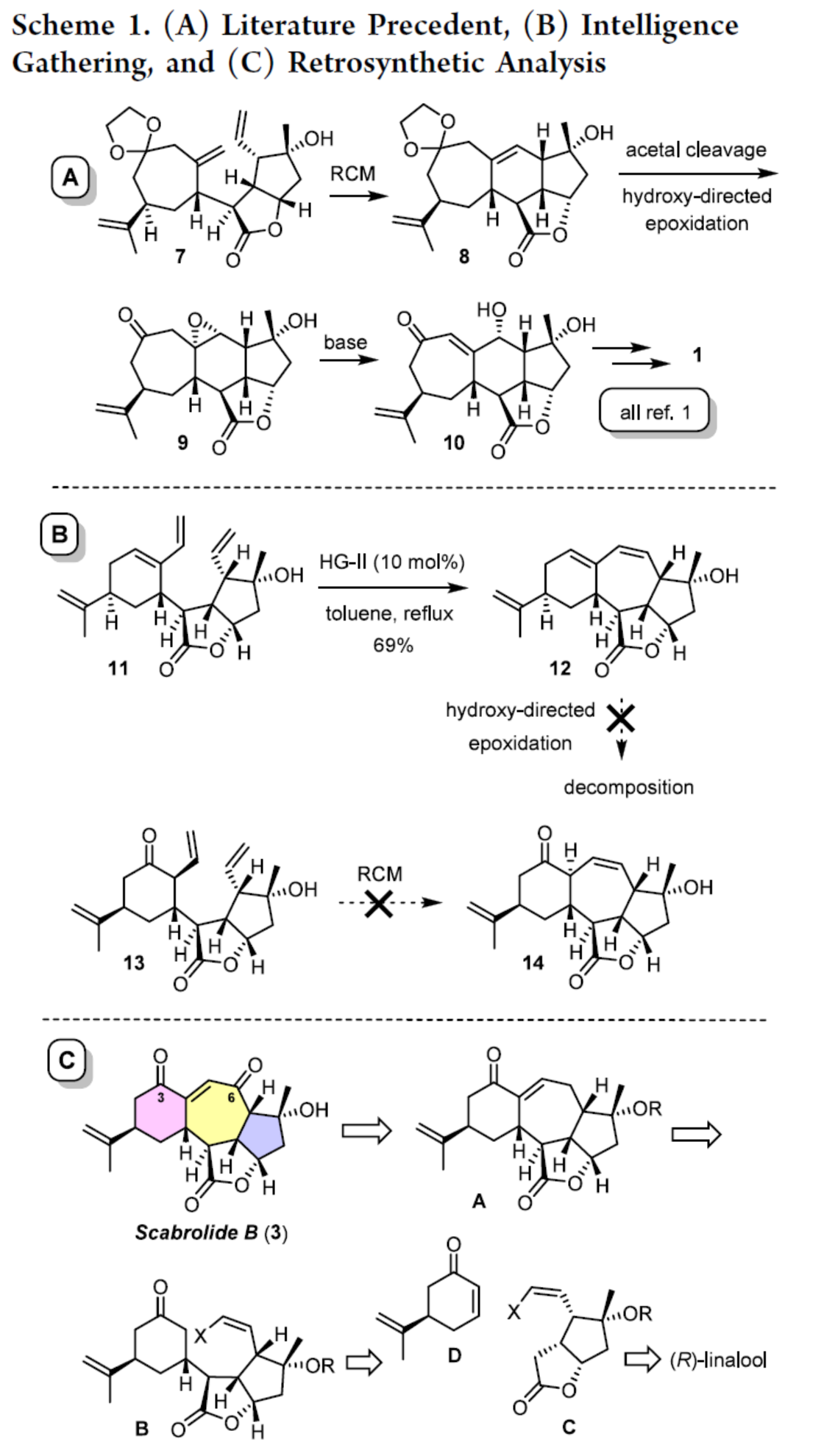
最初,作者对Scabrolide A(1)的合成路线进行一些调整,其也可能合成Scabrolide B(3)。具体来说,1的中心六元环是通过闭环复分解(RCM)构建。将所得烯烃化合物8进行羟基导向环氧化,然后进行碱诱导开环,以实现不协调的1,4-二氧化模式(Scheme 1A)。通过上述几个步骤,可将化合物10转化为目标产物。然而,这种策略不能延伸到Scabrolide B的合成(Scheme 1B)。虽然二烯化合物11能够顺利进行了闭环,但在12参与的羟基导向环氧化反应均失败。同时,化合物13通过RCM制备化合物14的尝试也未能成功。因此,作者进行了相关的逆合成分析(Scheme 1C)。首先,Scabrolide B(3)可由砌块A通过分子内烯醇化烯基化/氧化制备。其次,砌块A可由环化前体B制备。此外,环化前体B可由内酯化合物C与烯酮D通过Michael加成制备。
(图片来源:J. Am. Chem. Soc.)
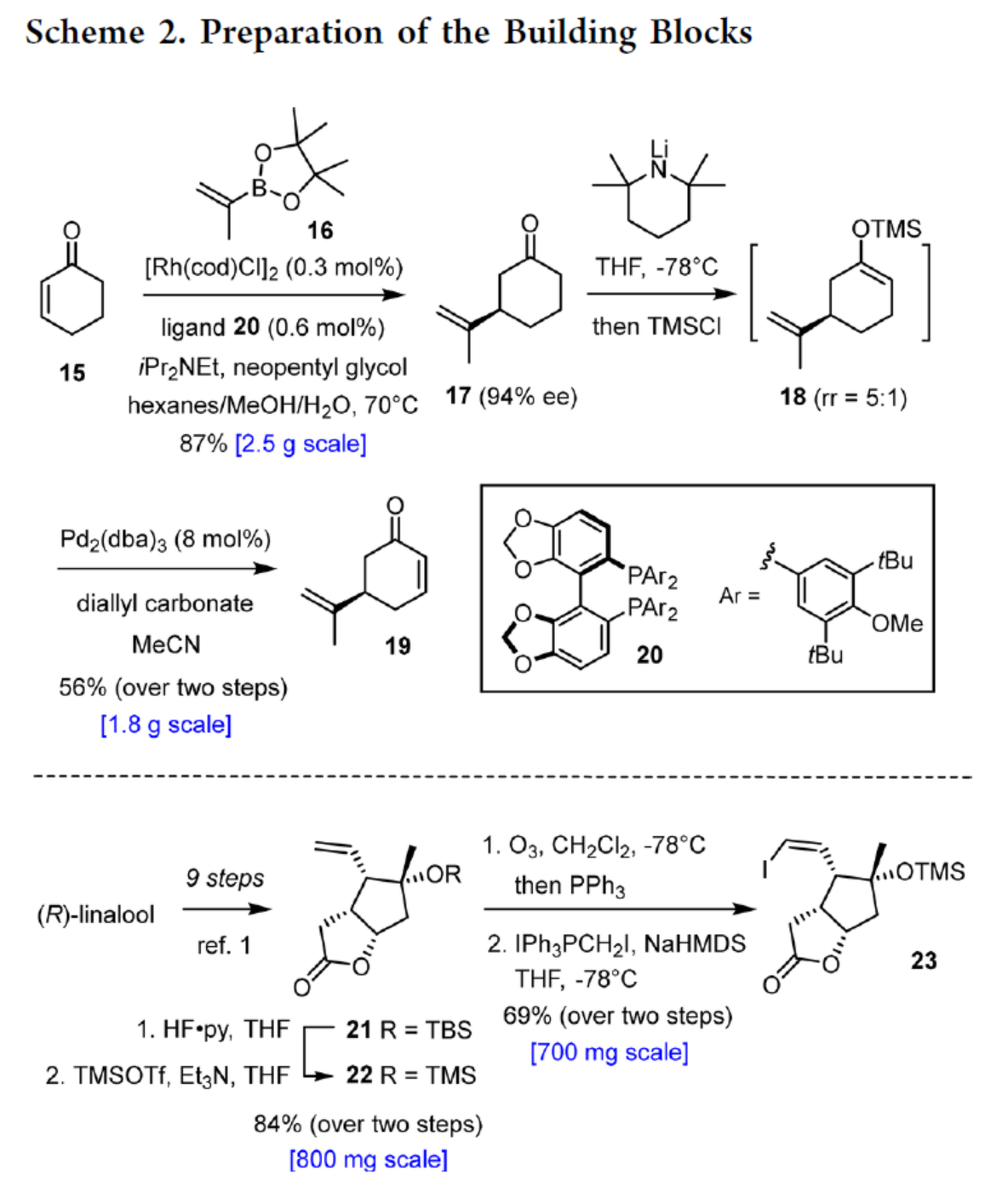
砌块19的合成(Scheme 2)。以环己烯酮(15)与硼酸酯(16)为底物,通过不对称铑催化1,4-加成反应,可以87%的收率得到中间体17,ee为94%。其次,使用位阻大的LiTMP进行脱质子化后,再使用TMSCl进行淬灭,生成硅基烯醇醚18作为主要异构体(rr ≥ 5:1,无需分离)。随后,以Pd2(dba)3为催化剂,碳酸二烯丙酯为终端氧化剂,无需任何额外配体,可直接进行Saegusa-型氧化反应,可以两步56%的总收率得到(R)-Norcarvone砌块19。
砌块23的合成(Scheme 2)。以(R)-芳樟醇为底物,按照文献的工艺(J. Am. Chem. Soc. 2022, 144, 1528; Chem. Rev. 2017, 117, 11753.),可制备中间体21。中间体21在HF·Py/THF与TMSOTf/Et3N/THF条件下进行保护基团替换,可以两步84%的总收率合成中间体22。中间体22通过进一步的臭氧化与Stork-Zhao烯基化反应,可以两步69%的总收率得到Z-烯基碘化物中间体23。
(图片来源:J. Am. Chem. Soc.)
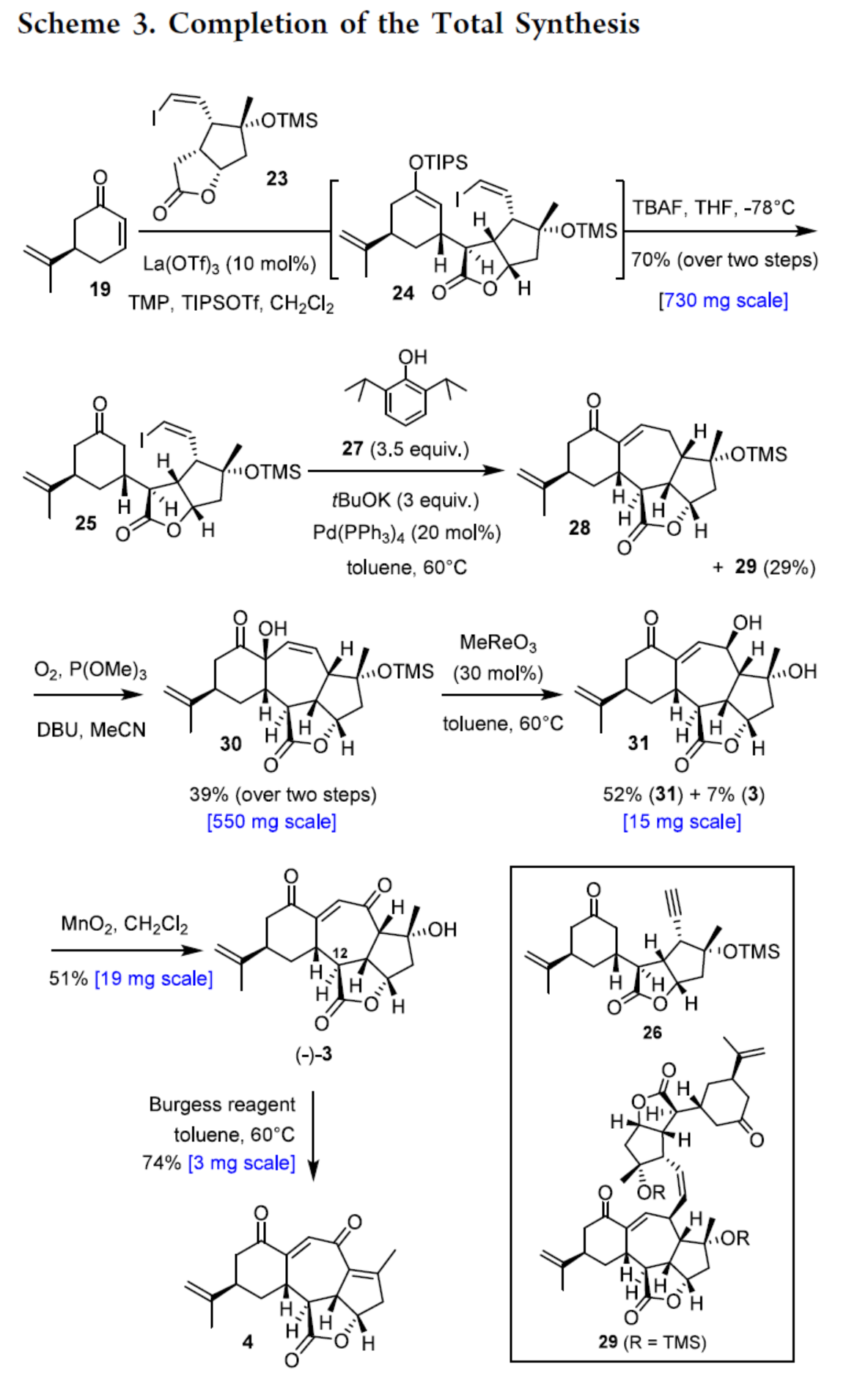
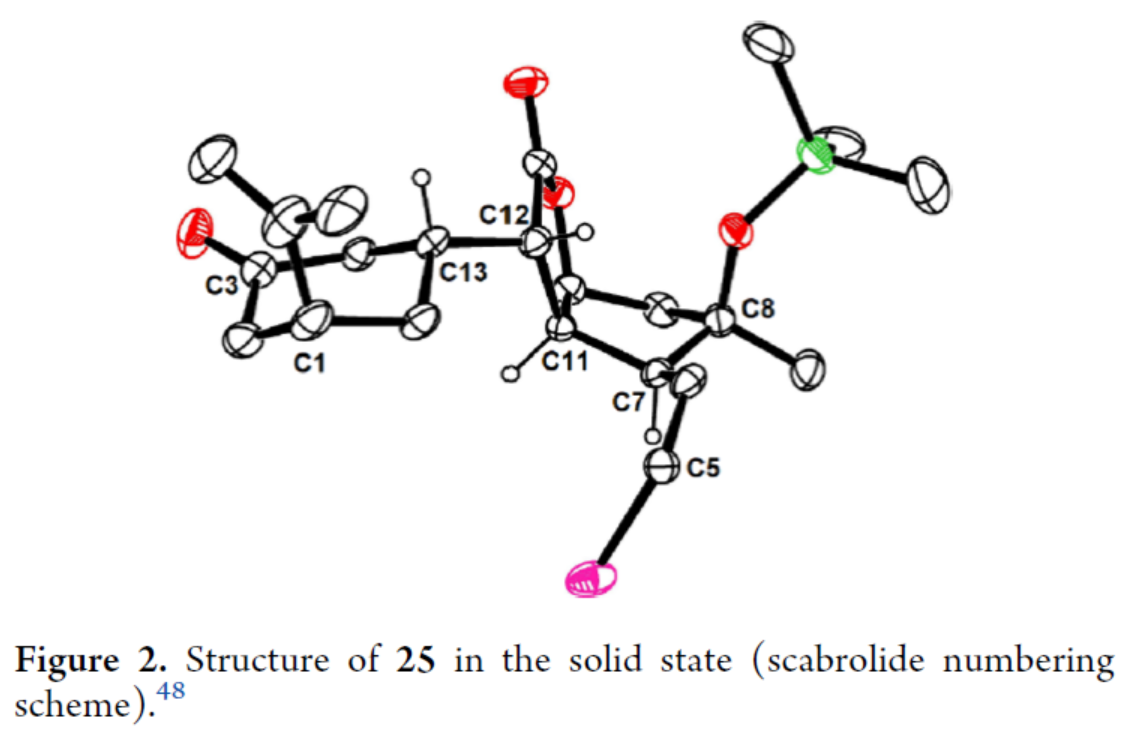
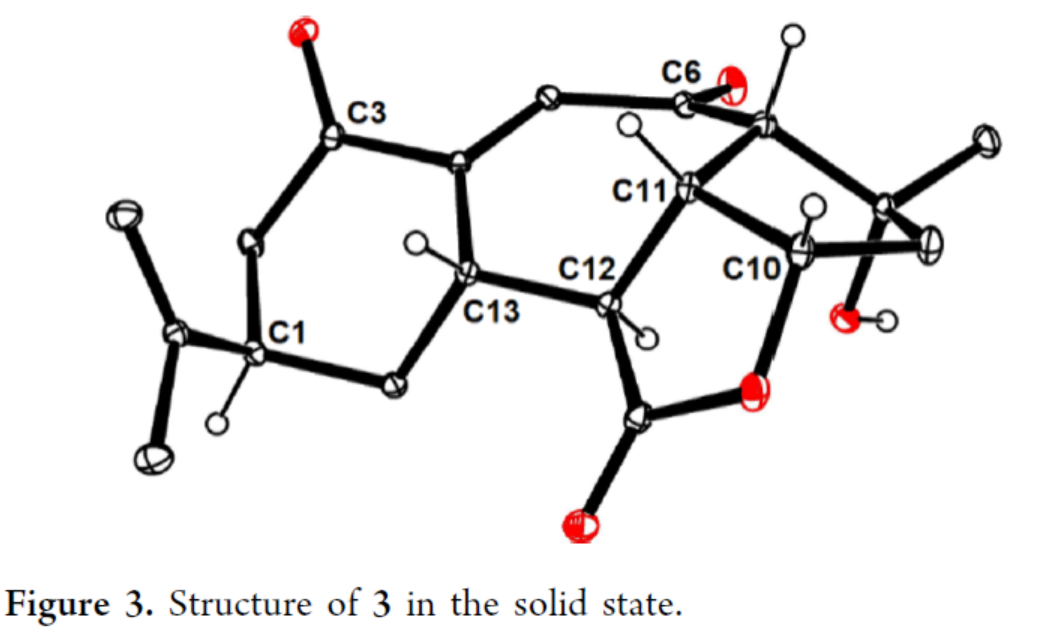
Scarrolid B(3)和Sinuscalide C(4)的全合成(Scheme 3)。以砌块19和砌块23为底物,在La(OTf)3/TMP/TIPSOTf条件下进行Mukaiyama-Michael加成反应,并在TBAF/THF条件下进行硅基烯醇醚的选择性断裂,可以两步70%的总收率合成中间体25。同时,通过X-射线单晶衍射分析,作者对位阻较大的C12-C13键上新形成的立体中心进行了确证(Figure 2)。通过对反应条件的大量尝试后发现,中间体25在2,6-二异丙基苯酚(27)/tBuOK/ Pd(PPh3)4条件下进行环化反应,可生成三环烯酮中间体28(收率约60%)和二聚体副产物29(收率为29%),无需进行进一步的分离。通过对反应条件的大量优化后发现,上述的混合物28和29在O2/P(OMe)3/ DBU条件下进行烯丙基重排/氧化反应,可以两步39%的总收率合成单一的非对映异构体30。中间体30在MeReO3条件下进行1,3-烯丙基重排反应,可以52%的收率得到中间体31。中间体31在MnO2/CH2Cl2条件下进行氧化反应,可以51%的收率得到(−)-3。同时,通过X-射线衍射分析,进一步证明了(−)-3结构的正确性(Figure 3)。(−)-3在Burgess试剂存在下进行脱水反应,可以74%的收率得到Sinuscalide C(4),表征数据和文献报道的一致。
(图片来源:J. Am. Chem. Soc.)
(图片来源:J. Am. Chem. Soc.)
(图片来源:J. Am. Chem. Soc.)
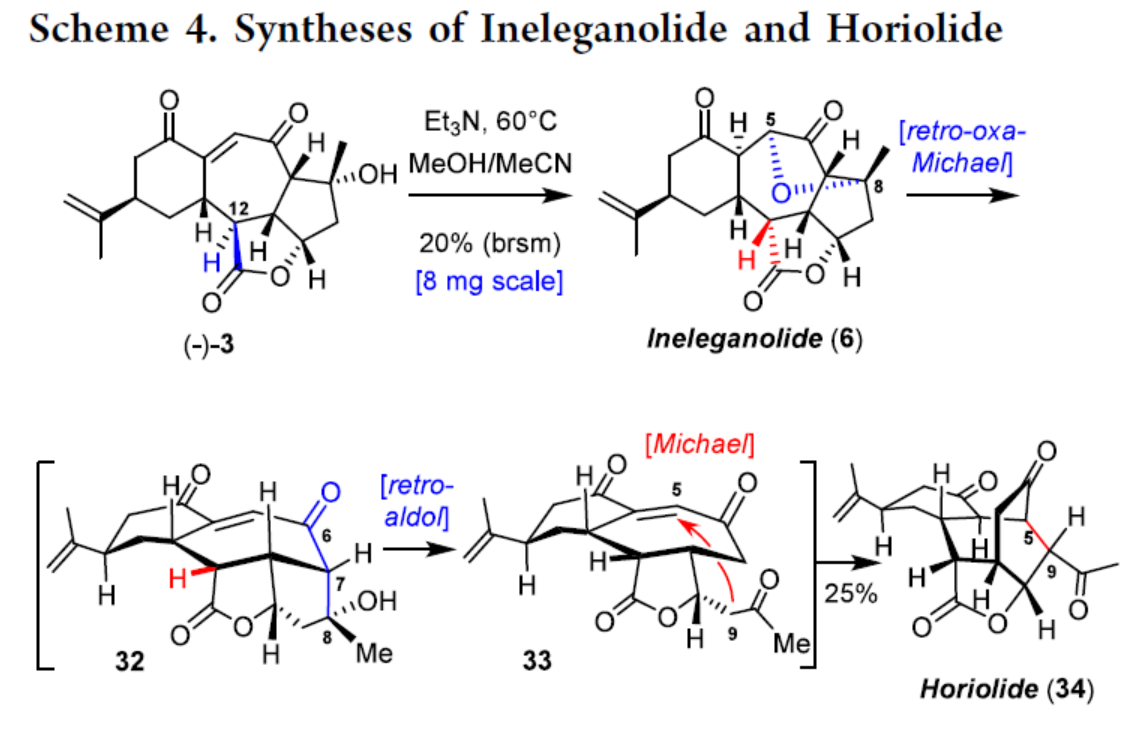
Ineleganolide(6)和Horiolide(34)的全合成(Scheme 4)。通过对反应条件的大量尝试后发现,以(−)-3为底物,在Et3N/MeOH/MeCN条件下进行串联反应,包括C8−OH基团在烯酮上的oxa-Michael加成反应和C12立体中心的差向异构化,可以20%的收率得到Ineleganolide(6)。Ineleganolide(6)通过进一步的retro-oxa-Michael反应,得到中间体32,32接着发生retro-aldol反应,得到中间体33,33再发生Michael加成,可以25%的收率得到Horiolide(34)。
(图片来源:J. Am. Chem. Soc.)
总结
德国马克斯普朗克煤炭研究所Alois Fürstner课题组报道了通过19步(最长线性序列)首次实现了Scabrolide B(3)的全合成,其可进一步转化为Sinuscalide C(4)、Ineleganolide(6)和Horiolide(34)。成功的关键是一种利用几乎对称的酮进行具有挑战性的分子内烯基化,这使得具有桥头烯烃的位阻大的七元环得以形成。虽然这种反应可以说是全新的,但显然具有很大的潜力。同样重要的是,Scabrolide B成功转化为Ineleganolide可能会模拟这些具有象征意义的海洋降西松烷类化合物之间的生源转化,值得进一步研究。
文献详情:
Total Synthesis of the Norcembranoid Scabrolide B and Its Transformation into Sinuscalide C, Ineleganolide, and Horiolide.
Davy S. Lin, Georg Späth, Zhanchao Meng, Lianne H. E. Wieske, Christophe Farès, Alois Fürstner*.
J. Am. Chem. Soc. 2024
https://doi.org/10.1021/jacs.4c09467

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国