

2020 年初,新冠病毒的阴影迅速笼罩全球。在这场与时间的赛跑中,我们见证了无数英勇的个体和团队挺身而出,社会体系经历了一次次严峻考验,也为全球的公共卫生领域敲响了警钟。
冠状病毒之所以令人畏惧,很大程度上是因为它属于 RNA 病毒。这类病毒在复制过程中缺乏纠错机制,容易发生突变。这种突变能力不仅允许 RNA 病毒跨物种传播,扩大宿主范围,还可能引发致病力的变化。原本对人类无害的病毒,一旦发生突变,就可能变得具有致病性,从而引发疾病。由于人类对这类突变病毒普遍缺乏免疫力,一旦病毒发生变异,就可能迅速引发大规模的疾病流行。
尽管病毒与人类健康密切相关,但人类已知已确认的病毒种类仅有 5,000 余种,可谓是冰山一角。传统 RNA 病毒鉴定方法高度依赖序列同源性比对,即通过比较未知病毒与已知病毒的序列相似性来进行识别。但是,由于 RNA 病毒种类繁多且高度分化,传统方法难以捕捉缺乏同源性或同源性极低的「暗物质病毒」,这限制了新病毒发现的效率。
在过去 10 年中,人工智能相关方法,尤其是深度学习算法,在生命科学领域的各个研究领域产生了重大影响。AI 与病毒学研究的结合,正在为人类突破 RNA 病毒鉴定这一难题提供新方法。
近日,**中山大学医学院的施莽教授联合浙江大学、复旦大学、中国农业大学、香港城市大学、广州大学、悉尼大学、阿里云飞天实验室等,提出了全新的深度学习模型 LucaProt。**该模型利用云计算与 AI 技术,发现了 180 个超群、16 万余种全新 RNA 病毒,是已知病毒种类的近 30 倍,大幅提升了业界对 RNA 病毒多样性和病毒演化历史的认知。该研究还发现了迄今为止最长的 RNA 病毒基因组,长度达到 47,250 个核苷酸,这标志着在 RNA 病毒鉴定领域取得了重大突破。
该研究以「Using artificial intelligence to document the hidden RNA virosphere」为题,发表在国际学术期刊 Cell 上。
研究亮点:
* 人工智能驱动的宏基因组挖掘技术,实现了全球 RNA 病毒多样性的空前扩展
* 通过精确鉴定,揭露了 161,979 种潜在 RNA 病毒物种和 180 个病毒超群的存在
* 该研究发现迄今为止最长的 RNA 病毒基因组,可能具有模块化的结构特征
数据集:涵盖全球各类生态系统, RNA 病毒存在多样性
该研究首先对 NCBI SRA、CNGBdb 等数据库进行了系统搜索,旨在对全球范围内各种生态系统中的 RNA 病毒多样性进行深入研究。
如下图 A 所示,研究团队总共从全球生物环境样本中筛选出来了 10,487 份数据,**涉及的总测序数据达到了 51 TB,产生了超过 13 亿个片段和 8.72 亿个预测蛋白。**利用这些庞大的数据集,研究者们揭示并验证了潜在的病毒 RdRP,并通过 2 种不同的策略进行了交叉验证。
RNA 病毒研究概述
通过合并 2 种搜索策略的结果,**该研究发现了 513,134 条病毒基因组,这些基因组代表了 161,979 个潜在的病毒种和 180 个 RNA 病毒超群。**这一发现显著扩展了该研究对 RNA 病毒超群数量的认识,将其扩容约 9 倍,病毒种类增加了约 30 倍。
如下图 C 所示,该研究通过与其他研究中的 RdRP 蛋白序列进行比较,共揭示了 70,458 个新识别的潜在独特病毒物种。

该研究的病毒超群分析
**该研究还揭示了 60 个以前未被识别的和未充分探索的超群,**这些超群迄今为止仅受到有限的关注。特别值得注意的是,如下图 D 所示,该研究发现其中 23 个超群无法通过传统的序列同源方法识别,这些被称为病毒圈的「暗物质」。

不同的 RNA 病毒簇和 RNA 病毒超群
LucaProt:基于数据驱动的深度学习模型,为病毒学研究开创全新范式
该研究开发了一个基于数据驱动的深度学习模型,即 LucaProt。如下图 E 所示,LucaProt 由 5 个核心模块组成:Input、Tokenizer、Encoder、Pooling和 Output:
* Input:主要负责接收氨基酸序列;
* Tokenizer:主要负责将原始序列转换为模型可以理解的格式。这一模块包括构建一个由病毒 RdRP 序列和非病毒 RdRP 序列组成的语料库,并使用 BPE 算法创建词汇表,将蛋白质序列分解为单个氨基酸以提取结构信息;
* Encoder:主要负责将数据转换为 2 种表示形式,一种是通过 Transformer-Encoder 生成的序列表示矩阵,另一种是通过结构预测模型 ESMFold 产生的结构表示矩阵。这种双轨表示方法既解决了 3D 结构数据稀缺的问题,又提高了计算效率;
* Pooling:主要负责通过值级注意力池化方法 (VLAP) 将序列矩阵和结构矩阵转换为 2 个向量,为有效分类减少维度并精选特征。
* Output:主要负责将这些向量转换成一个概率值,指示样本为病毒 RdRP 的可能性。通过 sigmoid 函数,将序列分类为病毒 RdRP 或非病毒 RdRP。
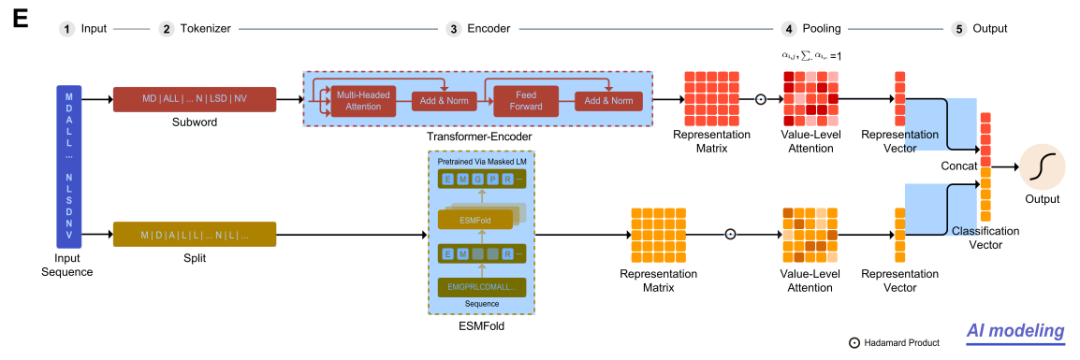
LucaProt 的 RdRP 识别方法
最终,**该研究精心筹备了包含 235,413 个样本的数据集,**旨在提升模型的准确性和泛化能力。这个数据集由 5,979 种已充分研究过的病毒 RdRP (阳性样本) 和 229,434 个非病毒 RdRP (阴性样本) 组成,基于 Transformer 框架与大模型表征技术,结合蛋白质序列和内在结构性特征,在准确性、效率和检测到的病毒多样性方面均优于传统方法。
更重要的是,LucaProt 不仅整合了序列数据,还结合了结构信息,这对于准确预测蛋白质功能至关重要。
识别出超出以往认知的基因组结构,发现迄今为止最长的 RNA 病毒基因组
为了全面评估 LucaProt 的性能,该研究进行了多角度的深入分析,以确保对其准确性和效率进行全面的验证:
* LucaProt 的效能评估
* 验证和确认新发现的病毒超群是否为 RNA 病毒
* RNA 病毒基因组结构的模块化和灵活性分析
* RNA 病毒系统发育多样性分析
* 全球 RNA 病毒的生态结构分析
5 种方法共同参与效能评估,LucaProt 的表现最为全面
为了评估 LucaProt 的效能,该研究将其与其他 4 种病毒发现工具进行了基准测试。结果表明,如图 A 所示,LucaProt 在保持相对较低的假阳性率的同时,展现出了最高的召回率。

召回率、精度和假阳性率分析
在计算效率方面,如图 E 所示,LucaProt 在处理不同长度的数据集时,基于 6 个数据集的平均计算时间,展现了更为合理的效率。

基于6个不同长度数据集计算的平均时间
最后,LucaProt 中集成的高级 Transformer 架构,允许对更长的氨基酸序列进行并行处理,如图 F-H 所示。这种架构在捕获序列空间中遥远部分之间的关系方面,比其它生物信息学工具中常用的 CNN/RNN 编码器更为有效。

基于测试数据集的预测结果比较
新发现RNA病毒超群的验证与结构特征确认,大多与现有 RdRP 表现出序列相似性
研究团队对 50 个环境样本进行了 DNA 和 RNA 的提取及测序,目的是验证这些样本中鉴定的 115 个病毒超群的存在。如图 B 所示,只有 RNA 测序读数能够成功映射到与病毒 RdRP 相关的序列,而 RNA 和 DNA 测序读数则分别映射到了与 DNA 病毒、逆转录病毒 (RT) 和细胞生物相关的序列。
进一步地,如图 C 所示,通过应用更灵敏的 RT-PCR 方法,研究团队在 115 个病毒超群中进一步确认了 17 个。在这些超群中,DNA 提取未能检测到编码病毒 RdRP 的序列,这进一步证实了这些病毒超群确实是 RNA 生物。

RNA 病毒超群的真实性评价
研究发现迄今为止最长的 RNA 病毒基因组
在深入分析假定 RNA 病毒基因组的组成和结构时,该研究发现尽管大多数基因组的长度集中在大约 2,131 个核苷酸,但编码 RdRP 的基因组或基因组片段在不同超群中长度差异显著。特别地,该研究从土壤样本中鉴定出极长的 RNA 病毒基因组,如图 C 所示,其中一条基因组长达 47.3 kb,**是目前已知最长的 RNA 病毒之一。**在这超长基因组中,该研究发现了一个额外的 ORF,位于第 50 端和 RdRP 编码区之间,但其功能尚需进一步研究。

病毒超群的基因组特征
RNA 病毒种的扩展速度惊人,环境样本中可能存在更多高度分化的 RNA 病毒
该研究还发现,如下图所示,与国际病毒分类委员会 (ICTV) 定义的病毒种相比,RNA 病毒种的数量增加了 55.9 倍,与所有先前描述的 RdRP 序列相比也增加了 1.4 倍。这种扩展在已知病毒群的多样性增加中尤为明显。

31 个 RNA 病毒超群的系统发育多样性分析
值得注意的是,一些之前仅由有限数量基因组代表的群体,例如 AstroPoty、Hypo、Yan 以及几个新发现的超群,展现出了高水平的系统发育多样性。例如,SG023 包含 1,232 种病毒,SG025 包含 466 种病毒,SG027 包含 475 种病毒。**这表明在环境样本中可能存在更多高度分化的RNA病毒,**等待我们去发现。
极端环境中的 RNA 病毒依旧存在多样性
**该研究显示 RNA 病毒遍布全球 1,612 个地点和 32 个生态系统。**如图 A 所示,即便在多次研究过的生态样本中,LucaProt 仍发现了 5-33.3% 的新病毒群,说明 RNA 病毒的多样性尚未充分挖掘,尤其是在土壤和水生环境。
研究还比较了不同生态系统中 RNA 病毒的 α 多样性和丰度。如图 C-D 所示,α 多样性在凋落叶、湿地、淡水和废水环境中最高,而丰度在南极沉积物、海洋沉积物和淡水生态系统中最高。岩盐和地下环境中的多样性和丰度最低,与宿主细胞数量少相符。极端生态亚型如温泉和热液喷口的 RNA 病毒多样性低,但丰度适中。
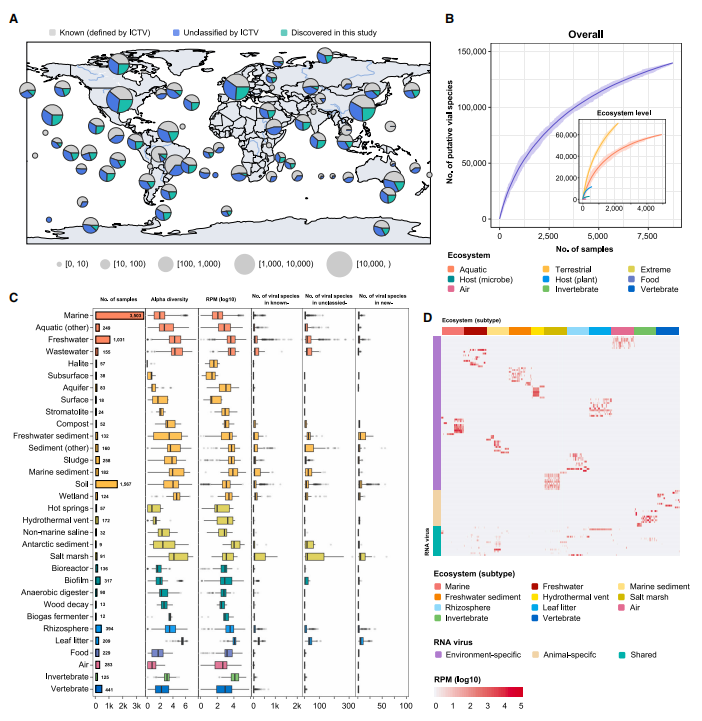
全球 RNA 病毒的生态结构
从学术到产业,AI 在 RNA 病毒研究中的革命性进展与未来展望
事实上,AI 在 RNA 病毒研究领域的应用已经成为科学探索的一股强大潮流。由中山大学施莽教授领衔的科研团队运用 AI 技术,取得了突破性进展,发现了超过 16 万种新的 RNA 病毒,这一成就标志着该领域的一个重要里程碑。
但早在 2022 年,一个国际研究团队在美国、法国、瑞士等国科学家的合作下,**就曾利用人工智能机器学习技术,从全球海水样本中鉴定出 5,500 种新的 RNA 病毒,**为RNA病毒数据库的建立做出了贡献。这项研究不仅拓宽了生态学研究的范畴,也加深了人们对 RNA 病毒进化的理解,为探索地球上早期生命的演化提供了新的线索。
该研究成果以「Cryptic and abundant marine viruses at the evolutionary origins of Earth’s RNA virome」为题已发表在 Science 杂志上。
当然,AI 在 RNA 病毒研究中的应用不仅限于未知领域的探索,对于已知领域的深入研究同样至关重要。例如,COVID-19 作为一种 RNA 病毒,其基因组序列在全球共享的 GISAID 数据库中已有近 1,600 万个。这些数据为研究提供了丰富的信息,但同时也需要大量的计算和人力资源来分析 COVID-19 的进化和历史。
为了应对这一挑战,2024 年初,曼彻斯特大学和牛津大学的科学家们开发了一种 AI 框架,该框架能够识别和追踪新的和相关的 COVID-19 变体,未来可能有助于解决其他感染问题。**这一框架结合了降维技术和曼彻斯特大学数学家开发的新型可解释聚类算法 CLASSIX,能够快速识别潜在风险的病毒基因组。**这项研究发表在「美国国家科学院院刊」上,为追踪病毒进化提供了新的方法,可能会对传统的病毒进化追踪方法产生影响。
在产业界,RNA 病毒研究的探索同样活跃。由于 RNA 病毒在复制过程中的高突变率,研究 RNA 病毒的疫苗开发一直是个难题。2023 年上半年,AI 辅助药物研发的应用日益增多,**百度加州分部的科学家利用 AI 对 mRNA 疫苗进行了深入的优化,不仅在序列上,还在结构上进行了改进,提高了分子的稳定性,**使其在人体内保持更长时间的活性。如果这一技术在安全性上得到验证,将成为新一代 RNA 疫苗研发的有力工具,也可能为 RNA 药物研发领域提供新的思路。
到了 2023 年下半年,Deep Genomics 公司发布了「An RNA foundation model enables discovery of disease mechanisms and candidate therapeutics」,介绍了其独特的人工智能基础模型 BigRNA。**BigRNA 是首个用于 RNA 生物学和治疗学的 Transformer 神经网络,拥有近 20 亿个可调参数,并在包含 1 万亿个基因组信号的数千个数据集上进行训练,**代表了新一代深度学习 AI,能够应用于多种不同的 RNA 治疗发现任务。
展望未来,AI 在 RNA 病毒研究中的应用前景也相当广阔。随着计算能力的提升和算法的改进,AI 或将能够处理更大规模的数据集,识别出更多未知的病毒种群,以及它们的宿主和传播途径。这不仅将加深人们对 RNA 病毒在生态系统中作用的理解,还将为预防和控制未来可能出现的疫情提供强有力的支持。
此外,AI 在疫苗设计和药物研发中的应用,预示着人们可能也即将迎来更加个性化和精准的医疗解决方案,为全球公共卫生安全带来了新的希望。


 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国