

导读
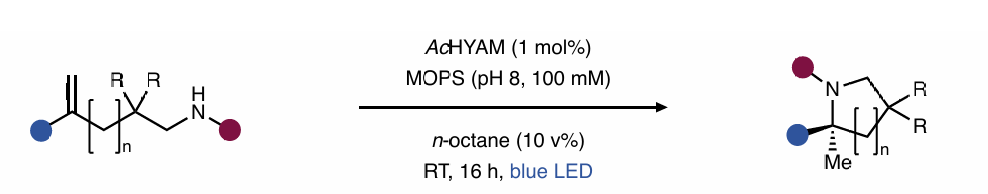
近日,美国普林斯顿大学(Princeton University)Todd K. Hyster课题组报道了一种光酶催化烯烃的氢-胺化反应,其通过Baeyer-Villiger单氧酶实现了一系列2,2-二取代吡咯烷的合成。作者通过五轮蛋白工程得到一个突变体,使得产物的产率和立体选择性都非常出色。与依赖胺基或烯烃氧化构建C-N键的相关光化学氢-胺化反应不同,此转化利用了还原生成的苄基自由基与氮原子的孤对电子之间的空间相互作用来构建C-N键。这种独特的C-N键构建机理在小分子催化中是难以实现的,为化学合成中未解决的挑战提供了独特的解决方案。
(图片来源:Nature)
正文
由于氮原子普遍存在于小分子药物和农用化学品中,因此发展高效的碳氮键构建方法在现代合成化学中至关重要。烯烃的氢-胺化是利用非活化烯烃构建碳氮键的一种原子经济的合成策略。然而,在制备全取代的碳立体中心时,这些反应很难实现不对称反应过程。最近,美国普林斯顿大学Todd K. Hyster课题组利用Baeyer-Villiger单氧酶实现了一种光酶催化烯烃的氢-胺化反应,构建了一系列2,2-二取代吡咯烷。通过五轮蛋白工程得到的蛋白突变体,可以以良好的产率和立体选择性得到产物。机理方面,此转化利用了还原生成的苄基自由基与氮原子的孤对电子之间的空间相互作用来构建C-N键,这与传统的利用胺基或烯烃氧化构建C-N键有所不同(Figure 1)。
(图片来源:Nature)
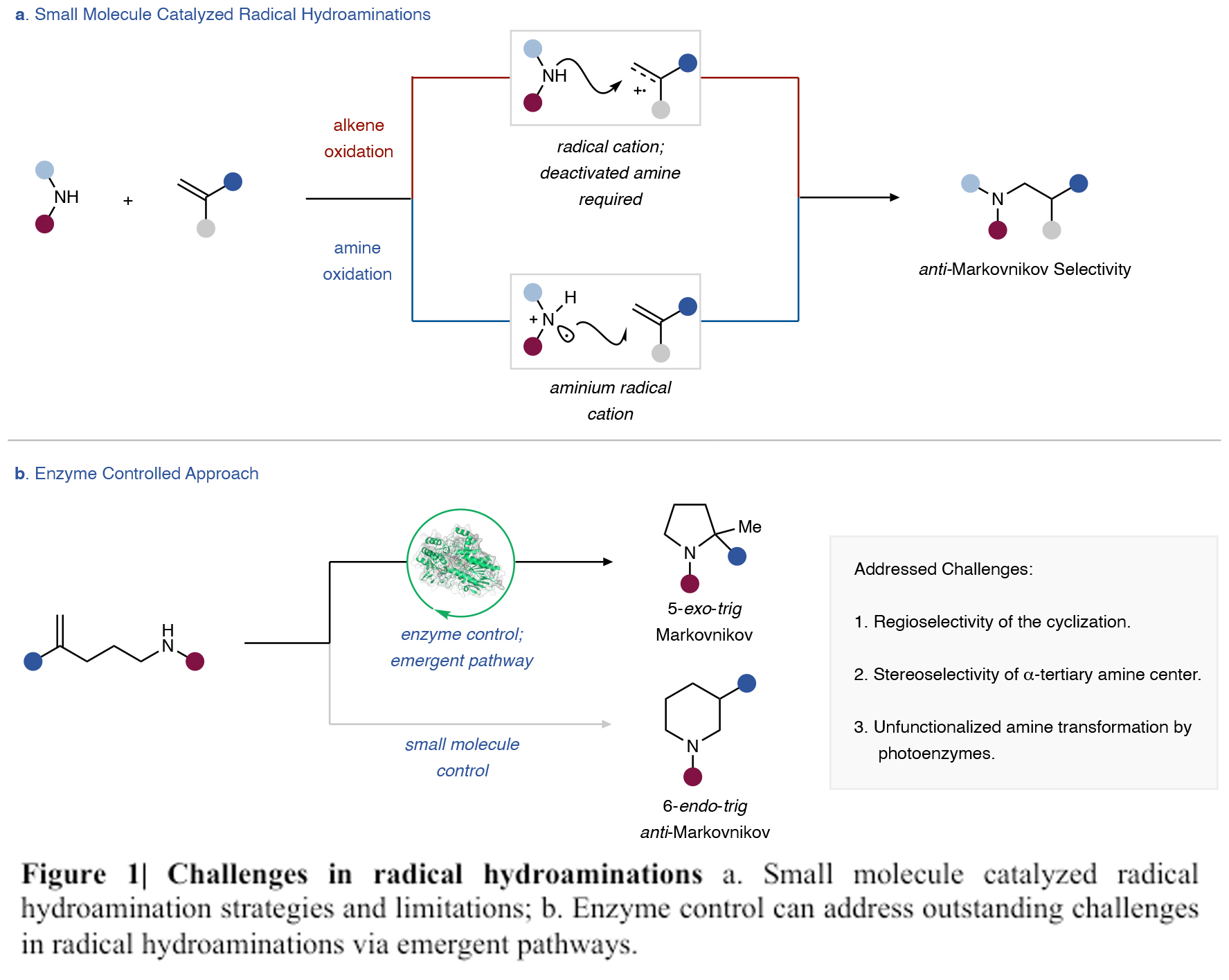
首先,作者以带有端烯的苯胺1为模板底物,探索了利用其构建2,2-双官能团化吡咯烷的转化(Figure 2)。通过对多种不同的黄素酶进行筛选,作者发现当使用AcCHMO(Acinetobacter calcoaceticus)时可以有效的实现5-exo-trig环化,以1%的产率,55:45 er得到吡咯烷产物2,且并没有观察到6-endo-trig产物3。而对于其它的酶,包括苯乙烯氧化酶、D-氨基酸氧化酶、单胺氧化酶、胆固醇氧化酶、氯氧化酶等均不能实现转化。由于利用蛋白质工程可以有效改善反应的产率和立体选择性,因此作者利用软件进行结构预测,将酶通过五轮蛋白工程得到一个突变体,其可以以97%的产率,95:5 er,选择性的得到吡咯烷产物2。分子动力学实验表明R490E、I490T和A479H三个位点位于一个环状区,是酶的活性位点。这种在远端位点突变的设计在打开催化活性位点和实现位点与底物的接触中起到至关重要的作用。随后,作者测试了无细胞状态下的酶催化活性。当将催化剂的载量降为0.04 mol %时,仍可以以17 %的产率得到产物4(TON = 436),且立体选择性得到保持。此外,作者还发现当加入1.0 equiv NADP+可以抑制反应,因此其可以作为有效的抑制剂。
(图片来源:Nature)
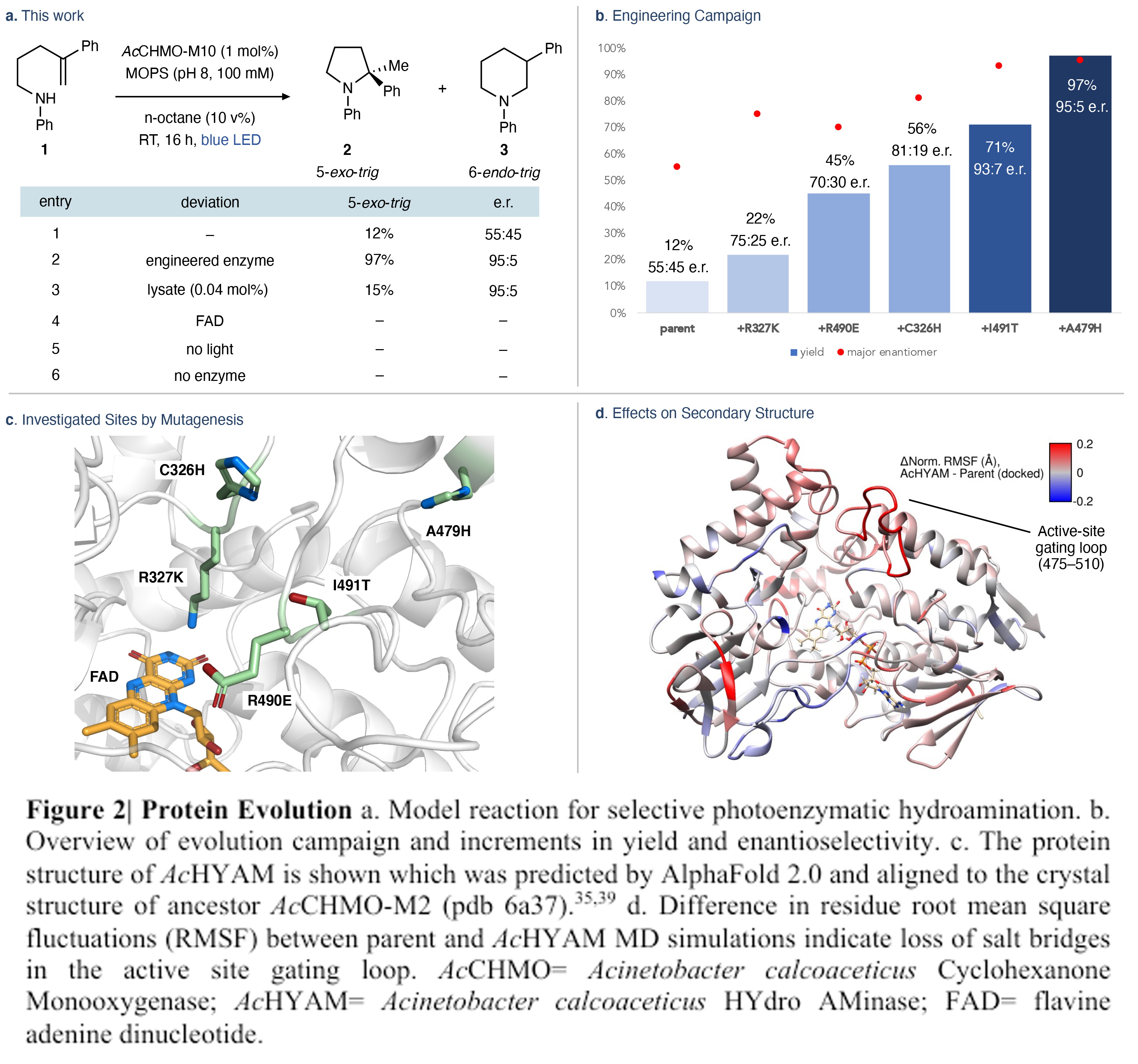
在最优反应条件下,作者对此转化的底物范围进行了考察(Figure 3)。实验结果表明,一系列不同取代的苯胺和苯乙烯均可良好兼容,以14-99%的产率得到相应的产物2-26。其中,富电子芳烃通常提供更高的产率和选择性(2、4-10、13-20)。苯胺一侧(10)和苯乙烯部分(19)上的大取代基,如萘,可以在没有降低产率和对映选择性的情况下进行转化。此外,缺电子的氮杂环同样是耐受的(11-12, 21),其克服了Brønsted酸催化环化的局限性。遗憾的是,环化反应对杂环取代较敏感,反应会受到影响。而利用蛋白质工程可以克服这些限制。相应的同系化底物可以实现6-exo-trig环化,以13%的产率得到产物22,并具有良好的对映选择性(78:22 er)。这是一个有趣的例子,因为当使用光氧化还原催化剂时,这种类型的底物通常会经历1,5-HAT而非环化。
此外,利用此策略还可以以中等产率(42%)和对映体比(27:73)得到修饰的吡咯烷骨架23。接下来,作者利用此方法制备了三种芳基吡咯烷原料药的α-甲基化类似物。包括TRK抑制剂GNF-862541的甲基化吡咯烷片段24(85%, 94:6 er);改良的原肌球蛋白抑制剂片段α-甲基larorectinib 26(77%, 75:25)和其相关结构25(25%, 73:27)。这些合成过程表明,利用此方法可以很容易的制备通常需要多步合成的取代类似物。
(图片来源:Nature)
接下来,作者对反应机理进行了探索(Figure 4)。作者发现该反应具有一定的诱导期,且不是由反应物的氧化来启动反应,诱导期的观察表明氧化黄素不是自由基引发的原因,而是需要光还原才能发生反应。而当使用连二亚硫酸钠还原酶进行催化反应时,在没有诱导期的情况下以74 %的产率,95:5 er得到产物。UV-Vis光谱表明,连二亚硫酸钠还原酶与光照还原酶是等效的。因此,此反应的诱导期是经历了光化学还原FAD为FADhq。此外,作者利用控制实验排除了能量转移机理的可能。基于上述实验结果,作者得出反应首先经历了烯烃被FADhq*还原。由于FADhq*的E0为-2.26 V vs. SCE,因此其无法还原α-甲基苯乙烯(Ered = -2.6 V vs. SCE)。但是正电性的活性位点可以减小反应物的还原电势,而且可以稳定FADsq-。这种阴离子具有很强的碱性,能够快速且不可逆的脱质子,从而形成苄基自由基。
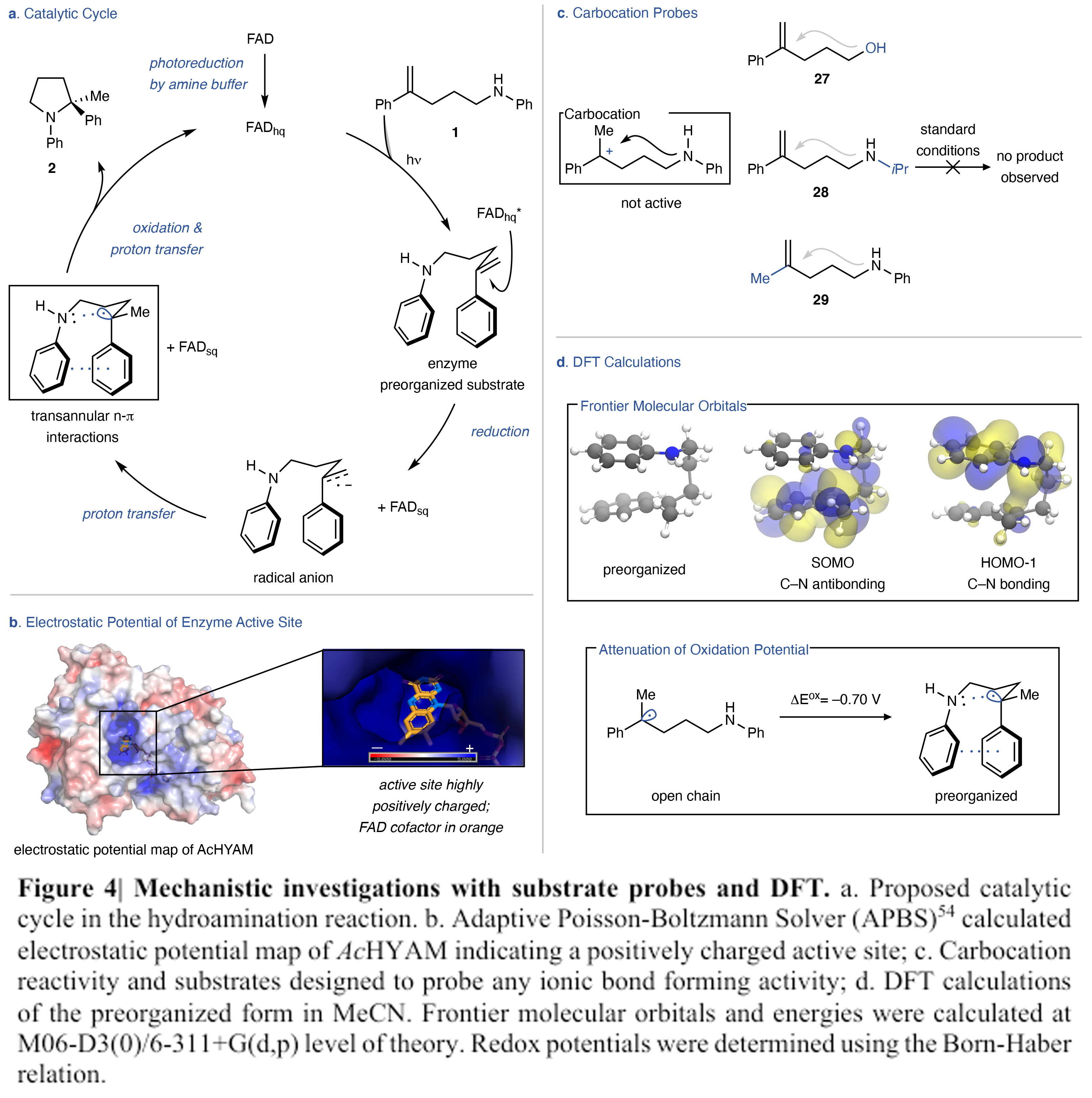
(图片来源:Nature)
作者通过研究发现,苄基自由基和苯胺之间的相互作用会影响氧化还原过程,且这种作用关系在以往的报道中还未出现。由于氮原子的孤对电子和自由基之间的超共轭作用,可以有效增加SOMO轨道的能量,使自由基更容易被氧化。因此此转化经历了一种与小分子催化完全不同的氢-胺基化机理。其利用苄基自由基与氮原子的孤对电子之间的空间相互作用来降低酶催化位点的自由基氧化还原电势从而构建C-N键。
总结
Todd K. Hyster课题组报道了一种高度选择性的光酶催化反应过程,立体选择性的实现了烯烃的氢-胺化反应。基于实验和计算结果,作者得出该反应经历了一种全新的C-N键形成机理,利用还原生成的苄基自由基与氮原子的孤对电子之间的空间相互作用实现了C-N键的构建。这种C-N键形成的新机理突出了利用酶实现新转化的潜力,其可有效解决化学合成中的挑战。
文献详情:
Emergence of a distinct mechanism of C–N bond formation in photoenzymes.
Felix C. Raps, Ariadna Rivas-Souchet, Chey M. Jones, Todd K. Hyster*.
Nature, 2024
https://doi.org/10.1038/s41586-024-08138-w.


 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国