

**LED、太阳能电池、光电探测器和光子集成电路 (PIC) 等光电设备是现代通信、照明和能源转换技术的核心。**这些设备的性能和效率在很大程度上取决于材料的光学特性,因此,深入理解这些特性对于推动技术进步和满足日益增长的科学及工业需求至关重要。为了应对这一挑战,实验和计算领域的研究人员通过积极开展高通量筛选工作,以寻找和开发具有定制光学特性的新型材料。
然而传统的获取材料光学特性的实验技术,如椭偏仪、紫外-可见光谱仪和傅立叶变换红外光谱仪 (FTIR),虽然能够提供精确的测量结果,但它们通常只适用于特定的波长范围,并且对样品条件有严格的要求。这些限制使得这些技术在高通量材料筛选中的应用受到了一定制约。
**为了解决这一问题,研究人员转向了基于密度泛函理论 (DFT) 的第一性原理计算。**与传统实验技术相比,DFT 计算能够覆盖所有波长范围的光学光谱,提供了一种更为全面的分析手段。尽管 DFT 计算能力强大,但在预测晶体结构的光学性质时,由于缺乏有效的原子嵌入,仍然面临着一定的挑战。
**针对于此,日本东北大学 (Tohoku University) 和麻省理工学院 (MIT) 的研究人员推出了一种新型人工智能工具 GNNOpt,成功识别出 246 种太阳能转换效率超过 32% 的材料,**以及 296 种具有高量子权重的量子材料,极大地加速了能源和量子材料的发现,为材料科学领域带来了新的研究范式。
相关研究以「Universal Ensemble-Embedding Graph Neural Network for Direct Prediction of Optical Spectra from Crystal Structures」为题,发表在 Advanced Materials 上。
研究亮点:
* GNNOpt 采用「集成嵌入」技术,不仅能够学习来自多种数据集的信息,还能够直接从晶体结构中精准预测所有线性光学光谱
* 通过集成等变神经网络,GNNOpt 利用 944 种材料组成的小型数据集实现了高质量的预测
* GNNOpt 从未知材料中成功出了筛选 246 种太阳能转换效率超过 32% 的材料,以及包括 SiOs 在内的 296 种具有高量子权重的量子材料

数据集:基于 944 种晶体材料的小样本学习
**研究人员使用基于密度泛函理论 (DFT) 计算得出的 944 种晶体材料,对 GNNOpt 模型进行光谱预测。**这些数据库是通过 API 从 Materials Project 获取得到的。而数据库中的光谱数据通过独立粒子近似 (IPA) 获取,包含了频率相关的介电函数及其对应的吸收系数。
整个数据集按照 80%、10% 和 10% 的比例,被随机分为训练集 (733 种材料)、验证集 (97 种材料) 和测试集 (110 种材料)。
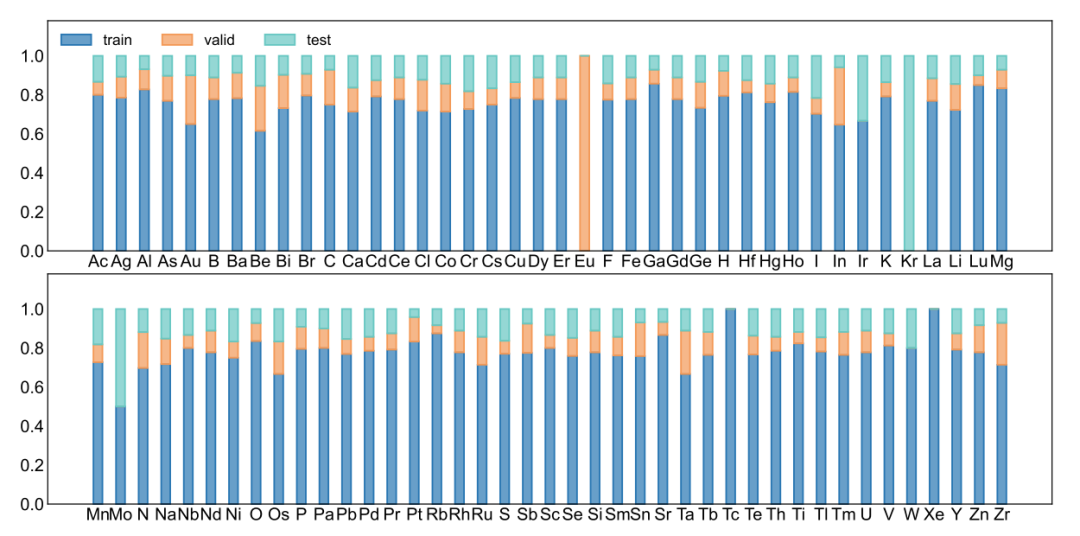
训练集、验证集、测试集元素分布
GNNOpt 模型架构:在晶体结构与频率相关光学特性之间直接建立关系
GNNOpt 是一种基于图神经网络 (GNN) 的模型,采用「集成嵌入」技术,能够直接从晶体结构中预测所有线性光学光谱。值得说明的是,在训练 GNNOpt 模型之前,研究人员通过一系列实验证明了应用克拉莫-克若尼关系式 (Kramers–Kronig relations) 能够更好地预测光学光谱。
如下图 a 所示,**GNNOpt 的唯一输入值是晶体结构 (Crystal structure),而输出值则是光谱,**具体包括复介电函数 (Complex dielectric function)、吸收系数 (Absorption coefficient)、复折射率 (Complex refractive index)、反折射率 (Reflectance)。
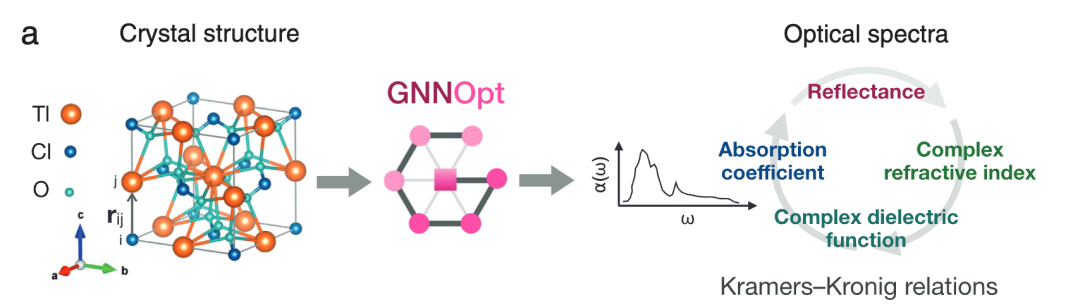
GNNOpt 模型的输入与输出示意图
在图 b 中,**晶体结构中每种原子种类 (O、CI、TI) 的输入特征采用独热编码表示。**由于元素周期表中的所有元素都有原子质量 (atomic mass, 用 x0 表示)、偶极子极化率 (dipole polarizability, 用 x1 表示) 和有效共价半径 (effective covalent radius, x2 表示) 这 3 种特征,因此研究人员选择这 3 个特征进行集成嵌入。
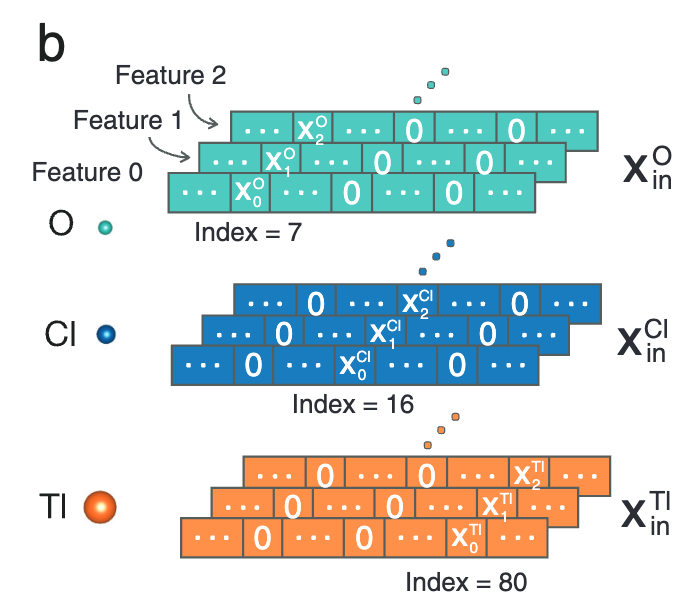
集成嵌入的 3 种特征
研究人员通过引入自动嵌入优化的集成嵌入层,可在不修改神经网络结构的情况下提高模型预测精度,具体过程如下图 c 所示。
首先,所有原子输入特征通过集成嵌入层 (Ensemble embedding) 进行自动优化。为了实现等变性,卷积滤波器由可学习的径向函数和球谐函数 (Spherical Harmonics) 组成。然后,将嵌入的特征通过一系列等变图卷积 (Graph convolution) 和门限非线性层 (Gated nonlinear layers) 参数化为可输入的参数。紧接着,参数结果被传递到包括激活和聚合操作的后处理层,用来生成预测的输出光谱。最后,通过最小化预测光谱与真实光谱之间的均方误差 (MSE) 损失函数来训练优化 GNNOpt 权重。
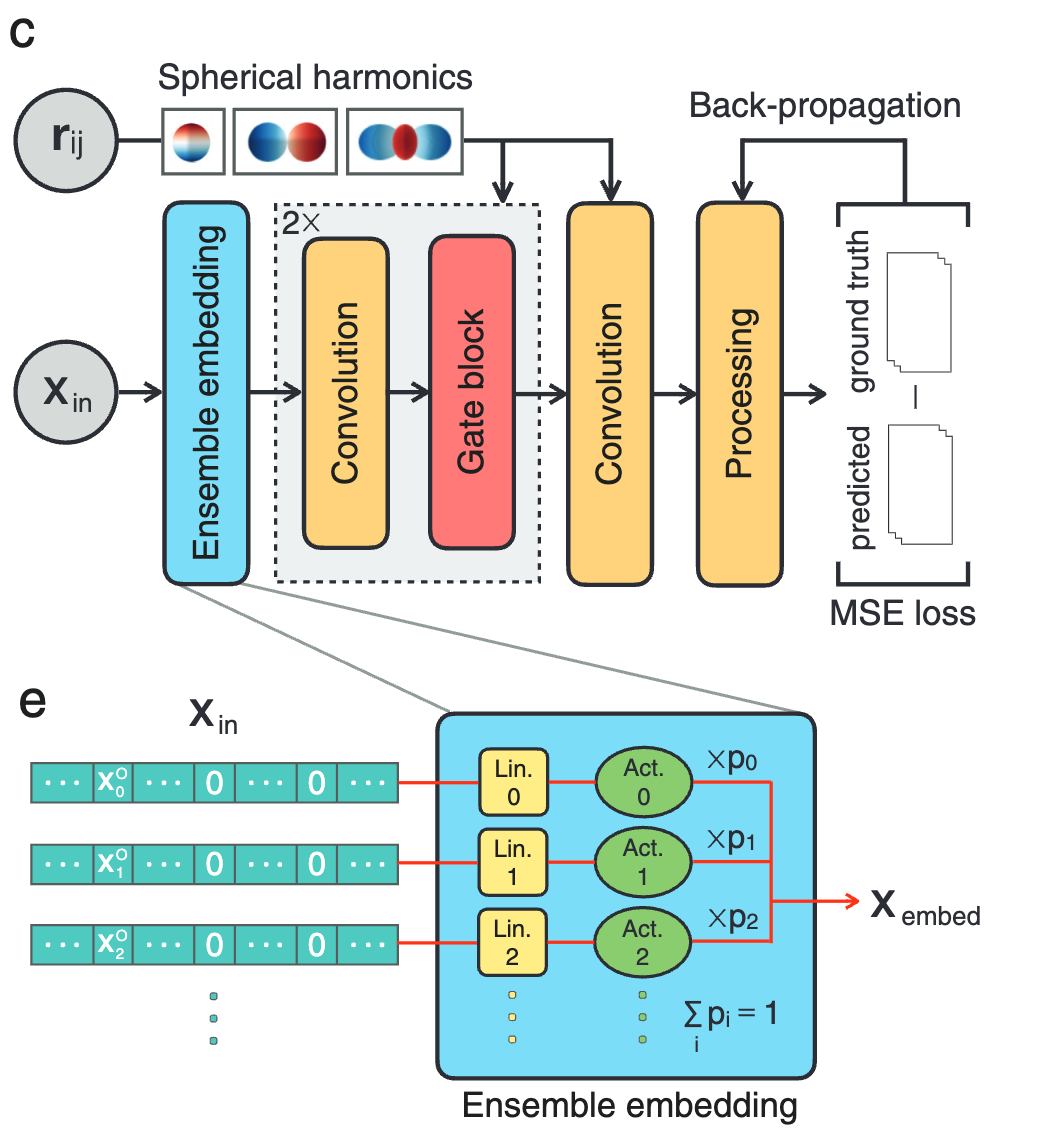
GNNOpt 模型架构示意图
**为了深入了解晶体结构,研究人员对 TlClO4 的晶胞 (unit cell) 结构进行了分析,如图 d 所示。**其中圆形节点代表晶胞内的原子,线条表示图卷积层的信息传递方向。
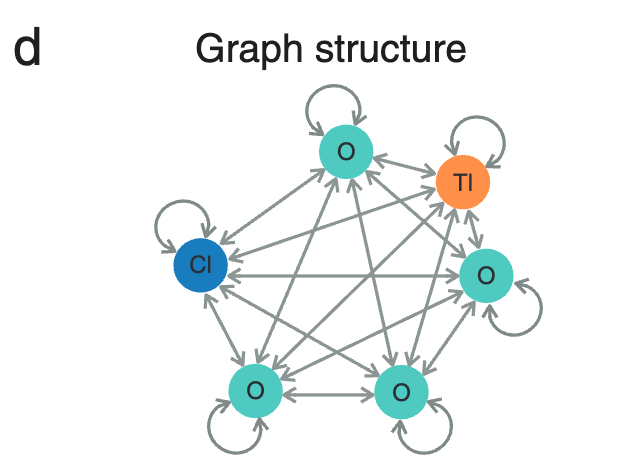
TlClO4 的晶胞结构
**图 e 则展示了通用集成嵌入层的细节,即使没有任何神经网络模型的改变,它也是性能提高的关键因素。**对于每个原子,每个特征都独立地嵌入其线性和活化层。然后,所有嵌入的特征通过可学习的混合概率 pi 进行加权平均,其中 pi 通过 ∑ipi = 1 进行归一化。
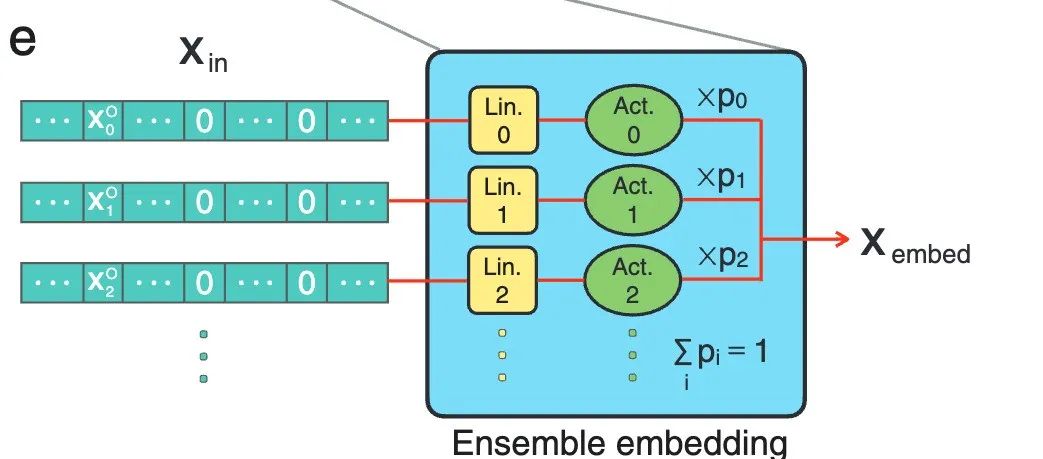
通用集成嵌入层细节展示
模型性能:GNNOpt 可识别出数百种太阳能电池以及量子候选材料
为了测试 GNNOpt 模型的性能,研究人员使用 GNNOpt 识别太阳能电池材料以及量子材料,成功识别出 246 种太阳能电池材料,以及 296 种具有高量子权重的量子材料。
上述材料的详细资料详见附加信息:
https://go.hyper.ai/rVSS8
关注公众号,后台回复「预测材料信息」获取完整 PDF
GNNOpt 能够从未知材料中筛选出 246 种太阳能电池材料
在识别具有高性能能量转换功能的潜在太阳能电池材料方面,研究人员使用光谱有限最大效率 (Spectroscopic Limited Maximum Efficiency,SLME) 方法,对太阳能电池的光电转换效率进行初步筛选和评估。
随后,研究人员使用 GNNOpt 模型预测了 Materials Project 中 5,281 种未知晶体结构的能量转换效率 (η 值),需要说明的是这些晶体结构没有真实的光谱数据。如下图 a 所示,研究人员比较了测试集的预测效率与真实效率,结果显示 R² = 0.81,表明 GNNOpt 对太阳能电池的光电转换效率的预测准确性较高。
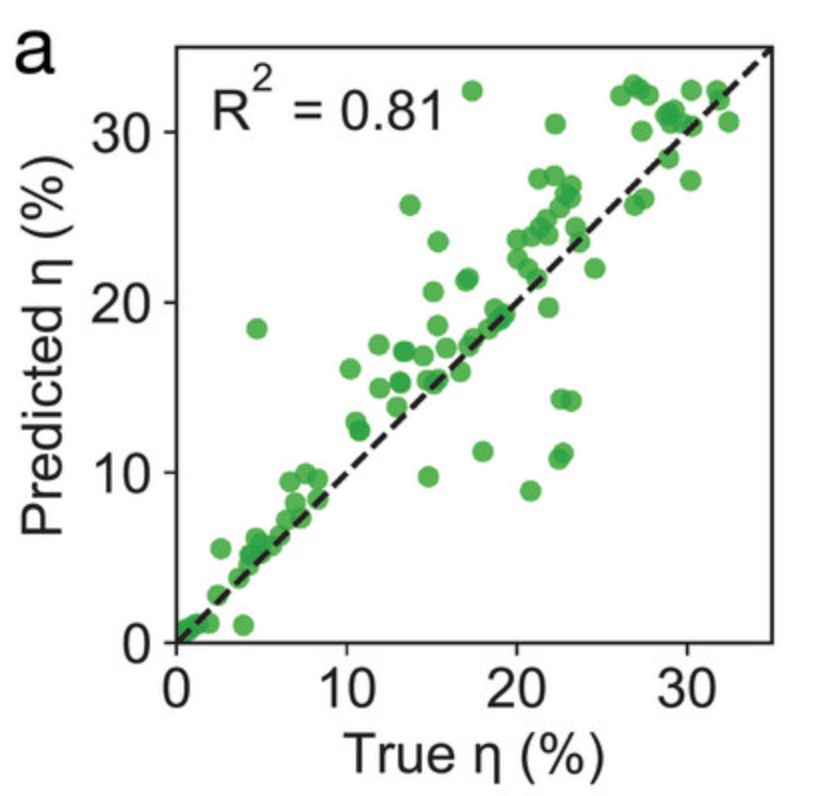
测试集中由 GNNOpt 预测的效率 η 与由 DFT 计算获得的真实效率 η 之间的对比
在图 b 中,研究人员将在测试集中由 GNNOpt 预测效率 η 与由 DFT 获得的真实效率 η 值,绘制成能量带隙 (energy band gap,用 Eg 表示) 的函数关系图。当 Eg 约为 1.3 eV 时,η 的最大值约为 32%,这与 SQ 极限一致。然而, SLME 作为太阳能电池材料的选择参数比 SQ 极限更加严格,因为对于具有相似带隙的材料,SLME 显示了 η 值在较宽范围内的变化,这表明吸收系数 α(E) 对 η 有着显著的贡献。
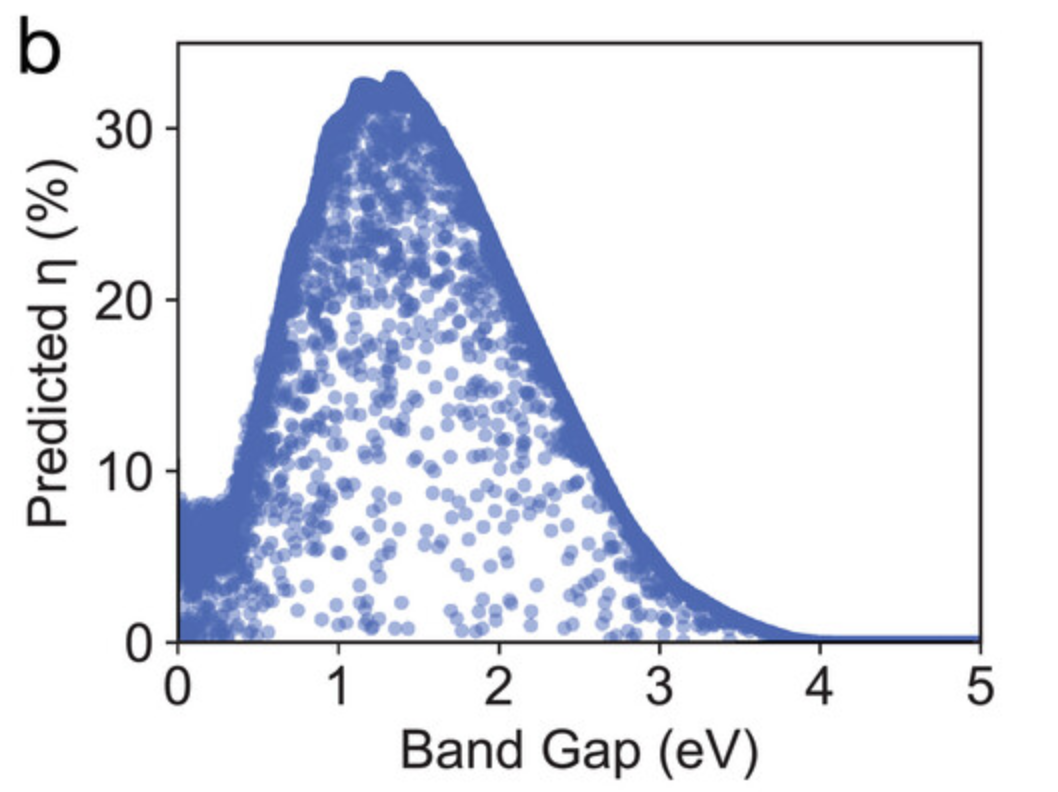
能量带隙函数关系图
此外,了解元素周期表中哪些元素对高效太阳能电池材料的贡献最大,可以为材料设计提供初步的指导。如图 c 所示,**GNNOpt 模型预测出过渡金属 (如 Tc、Rh、Pd、Pt、Cu、Ag、Au 和 Hg) 以及硫族元素 (如 S、Se 和 Te) 是太阳能电池材料的主要组成元素。**这一结果与广为人知的太阳能电池材料一致,如富铜的黄铜矿 (Cu-rich chalcopyrite)、铅基钙钛矿 (Pb-based perovskites) 或 CdTe。
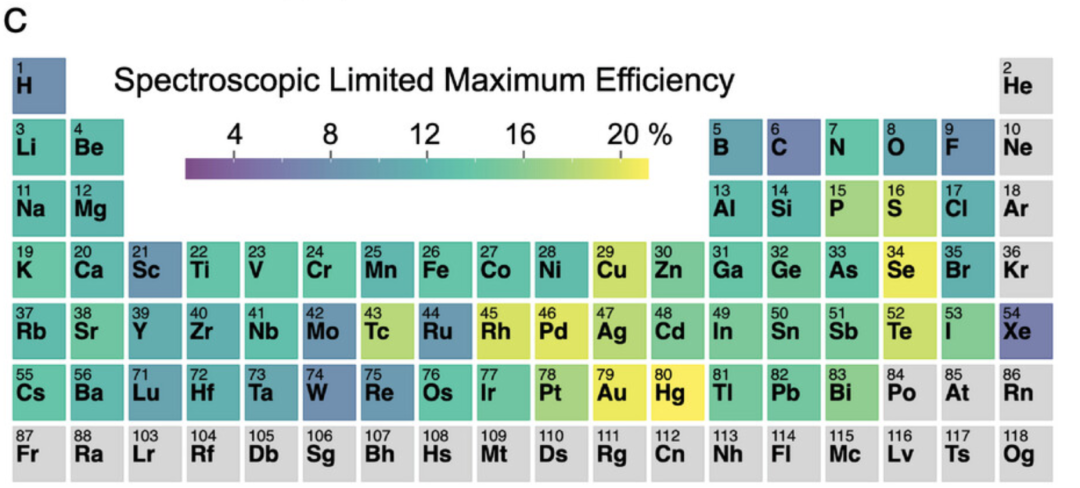
根据 SLME 对元素周期表进行着色
为了验证 GNNOpt 模型对未知材料的 SLME 预测值,研究人员从最高 SLME 材料列表中选取了 3 个示例:LiZnP、SbSeI 和 BiTeI。需要说明的是,这些材料不在 DFT 数据库中。因此,研究人员对这些材料进行了 DFT 计算,以确定这些材料的吸收系数 α(E)。
结果如下图 d 所示,DFT 计算的结果(用虚线表示)与 GNNOpt 预测的 α 值(用实线表示)高度吻合。这表明 GNNOpt 可以在大幅降低计算成本的情况下,成为一种有效的材料筛选工具。值得一提的是,对于大型数据库,可以将 GNNOpt 与遗传算法 (GA) 结合使用,从而加速候选材料的搜索过程。
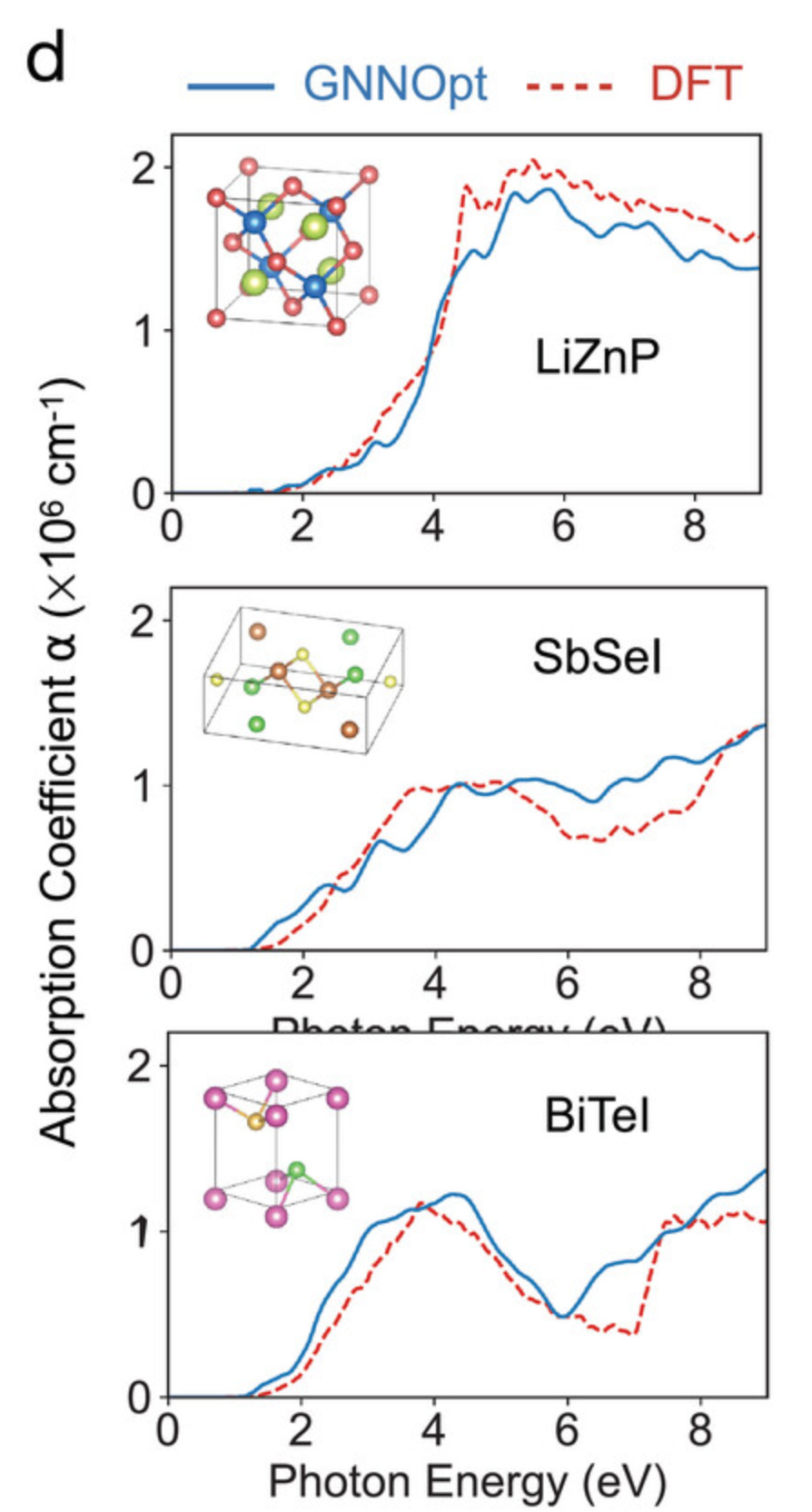
对于 3 种未知材料,GNNOpt 预测结果和 DFT 计算结果对比
GNNOpt 成功探测出包括 SiOs 在内的 296 种量子材料
除了可以识别未知的、具有高性能能量转换潜力的太阳能电池材料,**GNNOpt 的另一个应用是用于探测量子材料中的量子几何 (quantum geometry) 和拓扑结构 (topology)。**此前曾有学者表明,广义量子权重这一概念可以从光谱中推导出来,是衡量基态量子几何和拓扑结构的直接指标。量子权重 Kxx 是由反向频率加权的 f-sum 规则修正而来。
* 量子权重 Kxx 是量子系统中一个与材料的光学和电子性质相关的重要物理量,特别用来衡量其量子几何和拓扑特性。它描述了材料的量子几何结构与其光学性质或电学性质之间的关联。
在图 a 中,研究人员比较了测试集中以 h/e² 为单位的预测 Kxx 和真实 Kxx 的值。在 Kxx < 25 的范围内,R² = 0.73, 表明 GNNOpt 预测结果接近 DFT 计算的真实结果。
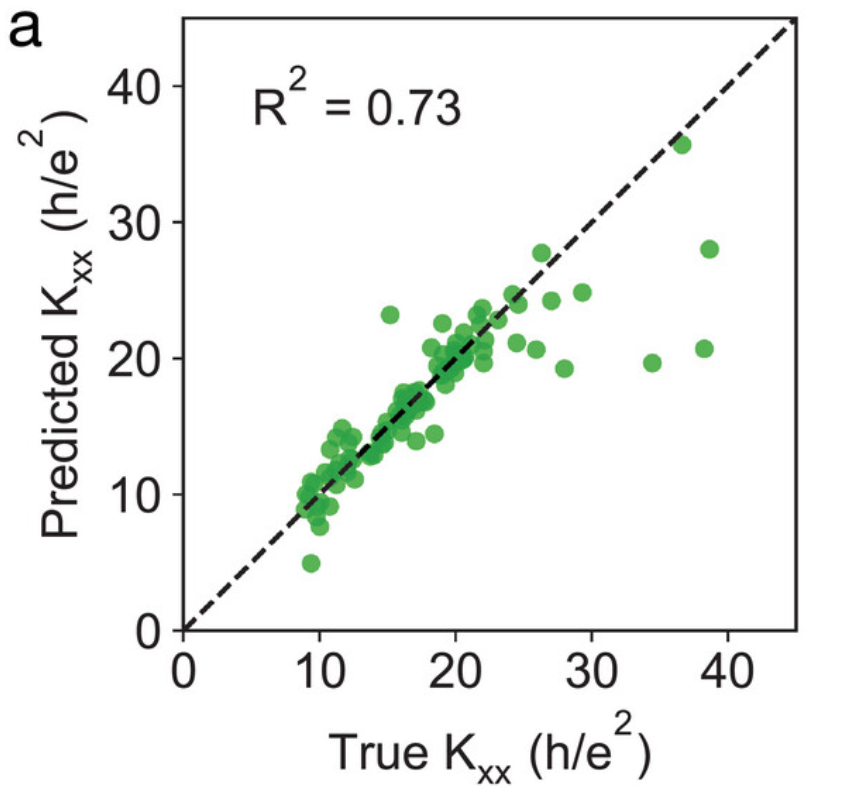
GNNOpt预测的Kxx和DFT计算的真实Kxx比较示意图
因此,GNNOpt 被用来预测 5,281 种未知绝缘体材料的 Kxx 值,如图 b 所示。为了简化分析,研究人员将著名的拓扑绝缘体 Bi2Te3 的量子权重 Kxx = 28.87 作为分类量子材料的阈值 (threshold),其中 Kxx > 28.87 的材料被视为高 Kxx 材料。
最终,研究人员确定了 297 种高 Kxx 材料。其中一些材料,如 ZrTe5 (Kxx = 33.90)、TaAs2 (Kxx = 37.66)、FeSi (Kxx = 48.74) 和 NbP (Kxx = 35.58) 等,已被确认为具有反常霍尔效应 (anomalous hall effect)、巨磁阻效应 (large magnetoresistance)、拓扑费米弧 (topological Fermi arcs) 和量子振荡 (quantum oscillations) 现象的量子材料。
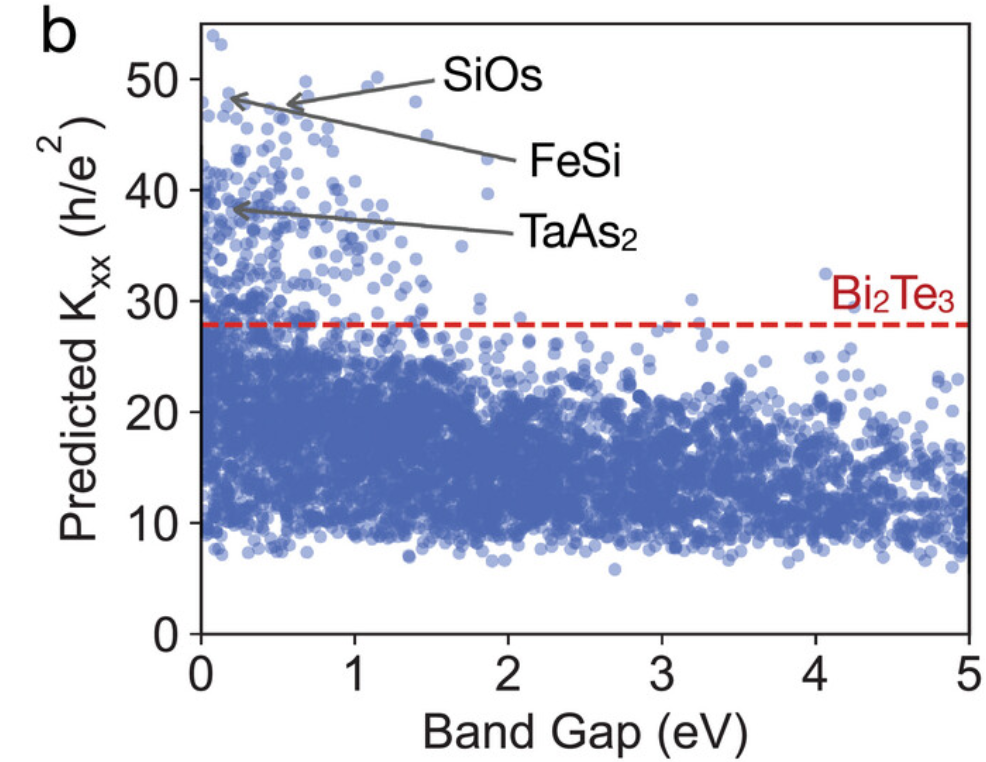
使用 GNNOpt 搜索具有高量子权重 Kxx 的量子材料
由于 SiOs 具有极高的量子权重 (Kxx = 46.52),且此前未被深入研究,因此研究人员对 SiOs 进行了额外的 DFT 计算,并分析了其电子能带结构 (Electronic band structure)。如图 c 所示,SiOs 在 Γ 点和 R 点分别存在三重费米子 (Three-fold fermion) 和双韦尔费米子(Double-Weyl fermion)。 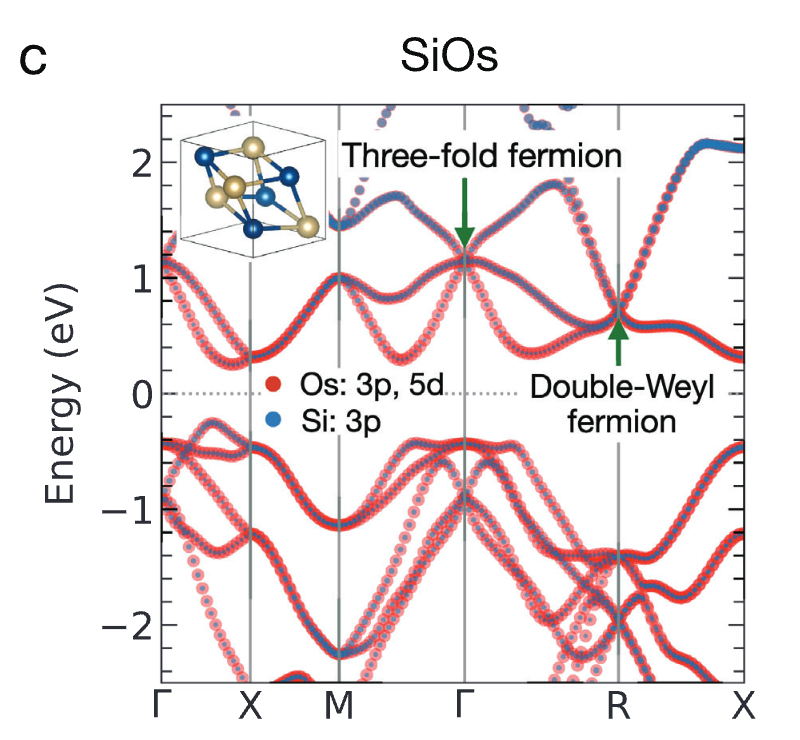
SiOs 电子能带结构
图 d 则是研究人员使用最大局域化万尼尔函数 (Wannier functions) 和格林函数 (Green’s function) 方法计算了 SiOs (001) 表面的能带结构,表明了 SiOs 的超量子特性。
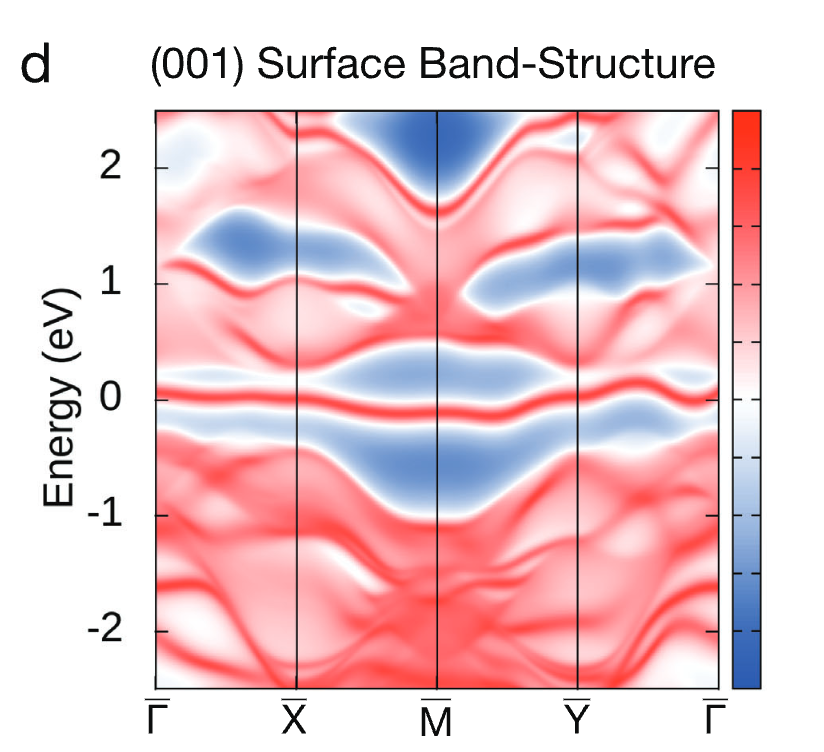
SiOs 电子能带结构的表面
人工智能将重塑材料研发过程,材料会逆向生成
在材料科学的快速发展中,AI 技术正在引领着一场革命。此前,中国工程院院士干勇曾公开表示,「人工智能将重塑材料研发过程,材料会逆向生成。」
首先,AI 在材料发现中的应用尤为显著。**2023 年 11 月底,谷歌旗下的 DeepMind 发布用于材料科学的 AI 强化学习模型 GNoME,**并通过该模型和高通量第一性原理 (DFT) 计算,寻找到了 38 万余个热力学稳定的晶体材料,极大加快了发现新材料的研究速度。
**微软不甘其后,在 GNoME 模型发表数天后,发布了材料科学领域的人工智能生成模型 MatterGen,**可根据所需要的材料性质按需预测新材料结构。
**2024 年 1 月,微软与美国能源部下属的西北太平洋国家实验室 (PNNL) 合作,利用人工智能和高性能计算,从 3,200 万种无机材料中筛选出了一种全固态电解质材料,**完成了从预测到实验的闭环,该技术可助力下一代锂离子电池材料研发。
此外,AI 也在材料性质预测方面发挥着重要作用。通过机器学习模型,可以预测材料的电子结构、力学性质等,从而优化材料设计。例如,**北京大学工学院研究员陈默涵开发的国产开源密度泛函理论软件 ABACUS,**结合 AI 辅助的交换关联泛函方法 DeePKS,克服了 DFT 计算在精度和效率上的两难问题,实现了高效率的杂化泛函精度计算。
AI 在材料科学领域的应用远不只此,在落地层面也涌现出诸如 Green Dynamics、CuspAl、DeepVerse 等致力于将 AI 运用于新材料领域的公司。随着技术的不断发展,AI 或将在材料科学领域迸发出无限的力量!

 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国