

导读
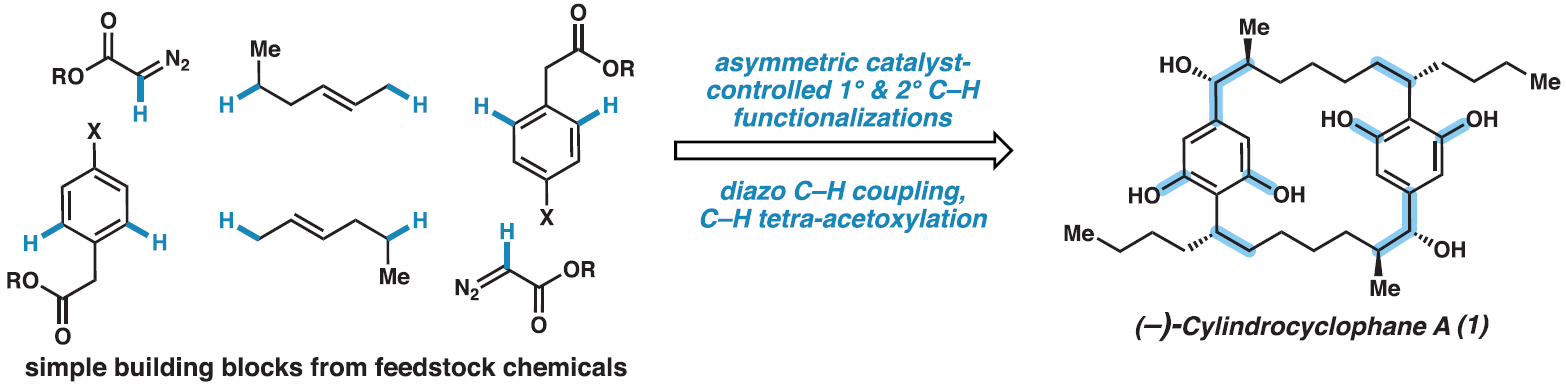
近日,美国埃默里大学(Emory University) Huw M. L. Davies课题组与美国加州理工学院(California Institute of Technology) Brian M. Stoltz****课题组联合报道了一种(−)-cylindrocyclophane A的合成策略。该策略借助10个C-H官能团化反应,以良好的对映选择性,共17步实现了(−)-cylindrocyclophane A的全合成。使用手性四羧酸二铑催化可以实现一级和二级C-H官能团化,其可以作为钯催化C(sp2)-C(sp2)交叉偶联的补充,实现大环核心骨架的构建以及具有高区域选择性、非对映选择性和对映选择性立体中心的快速构建。此外,利用后期钯催化的四重C(sp2)-H乙酰氧基化可以实现双间苯二酚部分的安装。
(图片来源:Science)
正文
C-H键广泛存在于药物合成的前体分子中,但它们通常是比较惰性的。在过去的二十年里,催化技术的快速发展使得C-H键的直接官能团化成为可能。(−)-Cylindrocyclophane A是一种22元C2-对称的[7.7]对二苯撑环烷烃,其具有双间苯二酚官能团和六个立体中心。而利用C-H官能团化策略实现(−)-cylindrocyclophane A的全合成则具有很大的挑战性。近日,美国埃默里大学Huw M. L. Davies课题组与美国加州理工学院Brian M. Stoltz课题组借助10个C-H官能团化反应,以良好的对映选择性和效率以17步实现了(−)-cylindrocyclophane A的全合成(Fig. 1)。
(图片来源:Science)
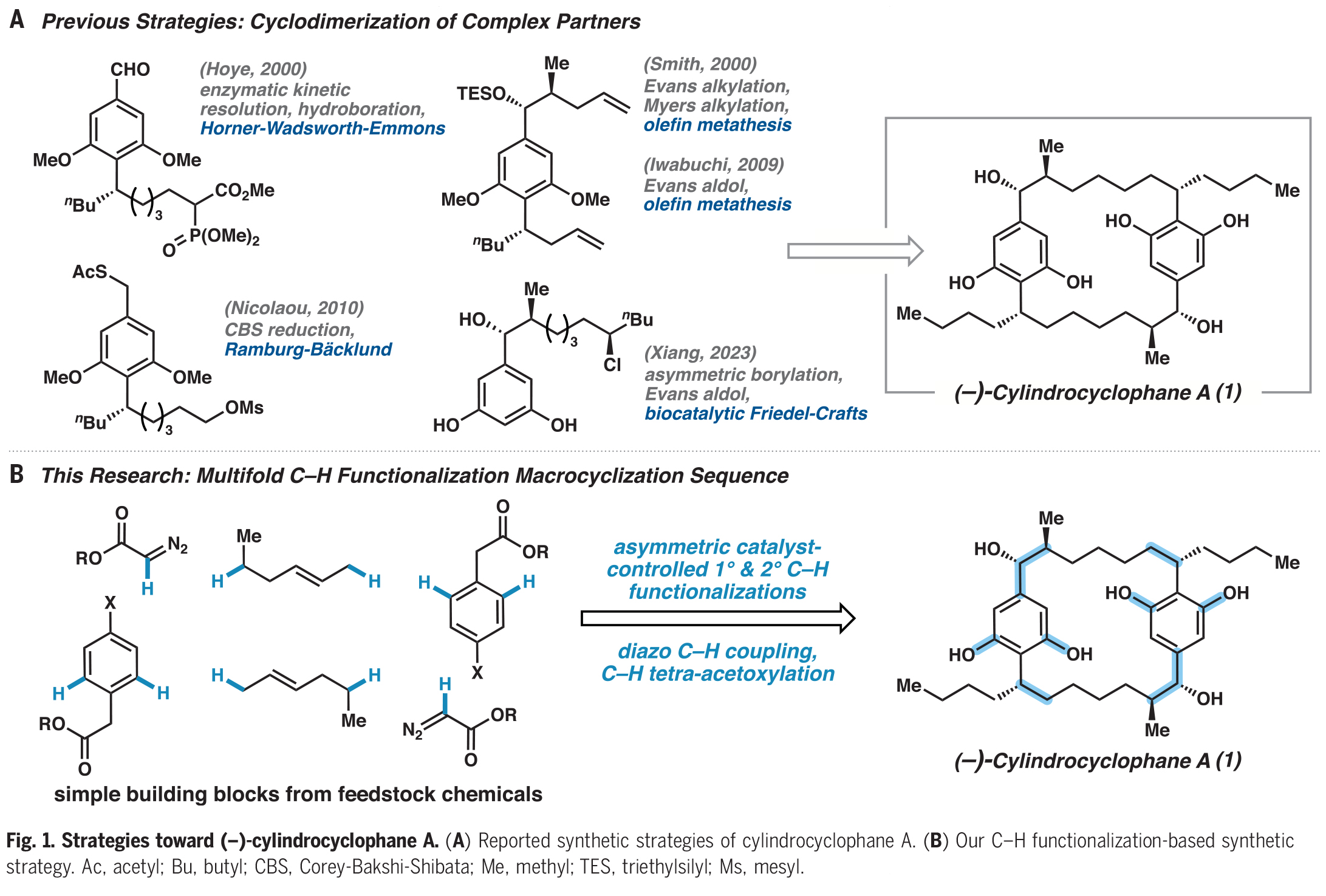
将C-H官能团化逻辑结合到(−)-cylindrocyclophane A的逆合成分析中,作者认为可以通过几种不同寻常的转化来快速简化分子(Fig. 2)。作者假设1可以通过双酰胺2中的每个Weinreb酰胺部分加入适当的亲核试剂得到。在后期通过二苯撑环烷烃3中四重Weinreb酰胺导向的C-H乙酰氧基化引入2,6-间苯二酚,从而降低分子的复杂性并调节了芳基的反应性。大环3中C1上的乙酸酯基和C7上的苄基酰胺可以逆推到一个正交保护的大环四酯化合物4。作者发现利用铑(II)催化的二级C(sp3)-H官能团化可以在天然产物中构建邻近的立体中心,并作为C-C键构建和大环化的关键策略。这些转化可以得到重氮酯5,其可以由钯催化重氮乙酸酯与芳基卤化物6的C-H官能团化反应来制备。最后,作者认为芳基卤化物6可以由铑(II)催化伯胺的C−H官能团化反应得到。
(图片来源:Science)
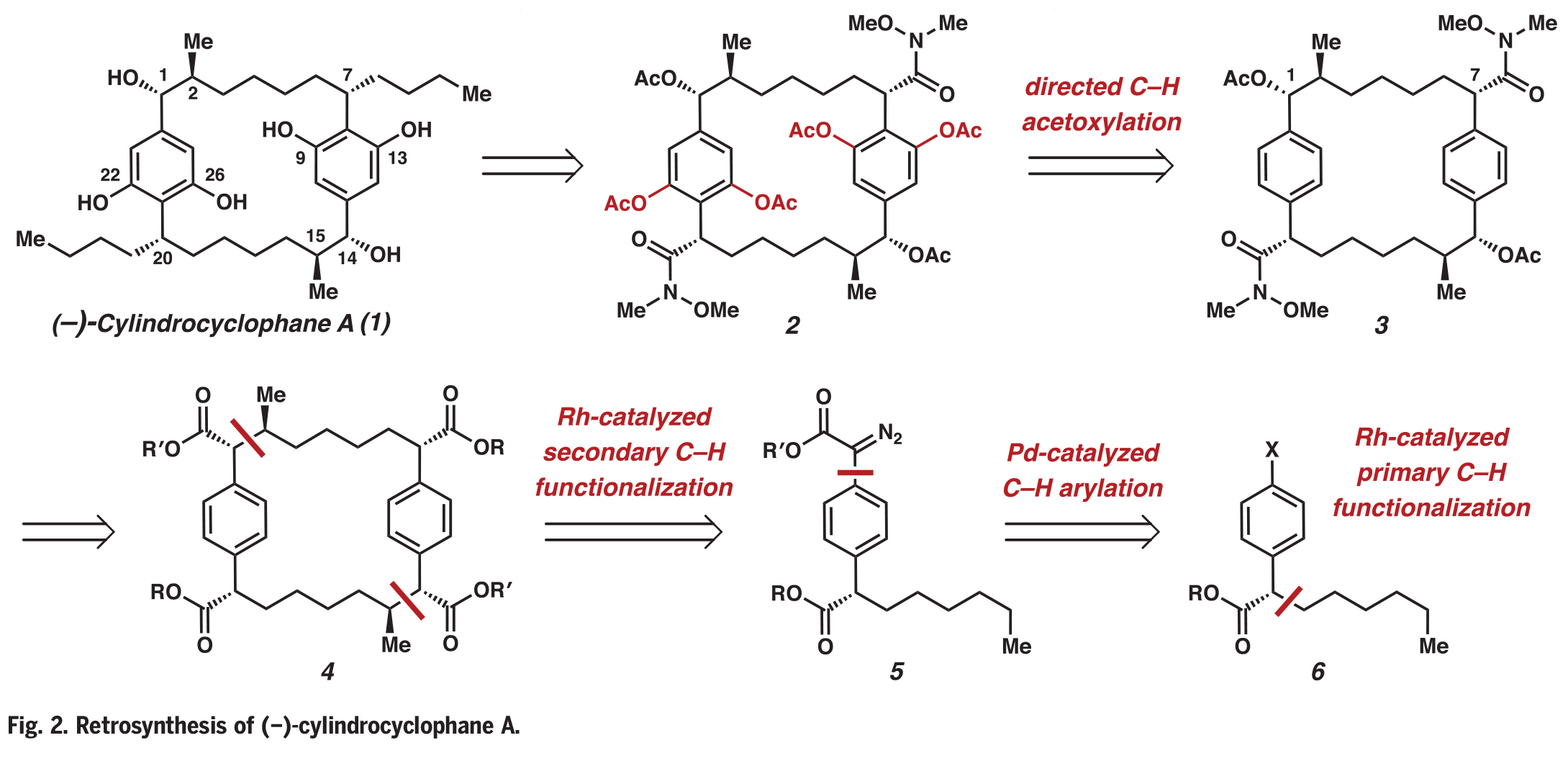
合成过程如下:首先,作者利用已报道的芳基重氮乙酸酯7和反式2-己烯8进行选择性一级碳氢官能团化(Fig. 3A)。当使用具有立体位阻的Rh2(R-p-PhTPCP)4催化剂时,该反应具有较高的区域选择性和对映选择性。该催化剂可以在较小立体位阻的C-H键上进行反应,以73%的产率、96% ee在C20上实现官能团化。使用Crabtree催化剂进行反式烯烃9的加氢反应可以以定量产率得到碘化物10。作者还试图通过正己烷的官能化直接获得10,但是此过程得到了分别经历伯和仲C-H官能化反应的不可分离的混合物。将碘化物10与重氮酯11进行钯催化的C(sp2)-C(sp2)交叉偶联,可以以88%的产率得到芳基重氮乙酸酯12。接下来,作者将注意力集中到C−H官能团化过程,从而构建[7.7]对二苯撑环烷烃骨架。
接下来,作者利用芳基重氮乙酸酯12,Rh2(R-2-Cl-5-BrTPCP)4和三当量的10生成的芳基碘化物13(产率68%,dr = 19:1)。双芳烃中间体13通过C-H官能团化又进行了一次钯催化的交叉偶联,以77%的产率得到了大环化前体14。用Rh2(R-2-Cl-5-BrTPCP)4与重氮酯14发生大环化过程,可以以70%的产率生成大环15的两个立体对映体(C14-C15)。此转化具有很高的不对称诱导效果(> 30:1 dr),但具有较低的非对映控制(8:1 dr)。与芳基碘化物13的形成相比,大环化的总体非对映选择性较低的原因是由于Horeau效应,其中不完全的不对称诱导产生的是非对映体混合物而不是对映体混合物。此外,此过程单次可单次制备> 1.2 mmol的大环15,并可以通过重结晶分离为单一的非对映体。
(图片来源:Science)
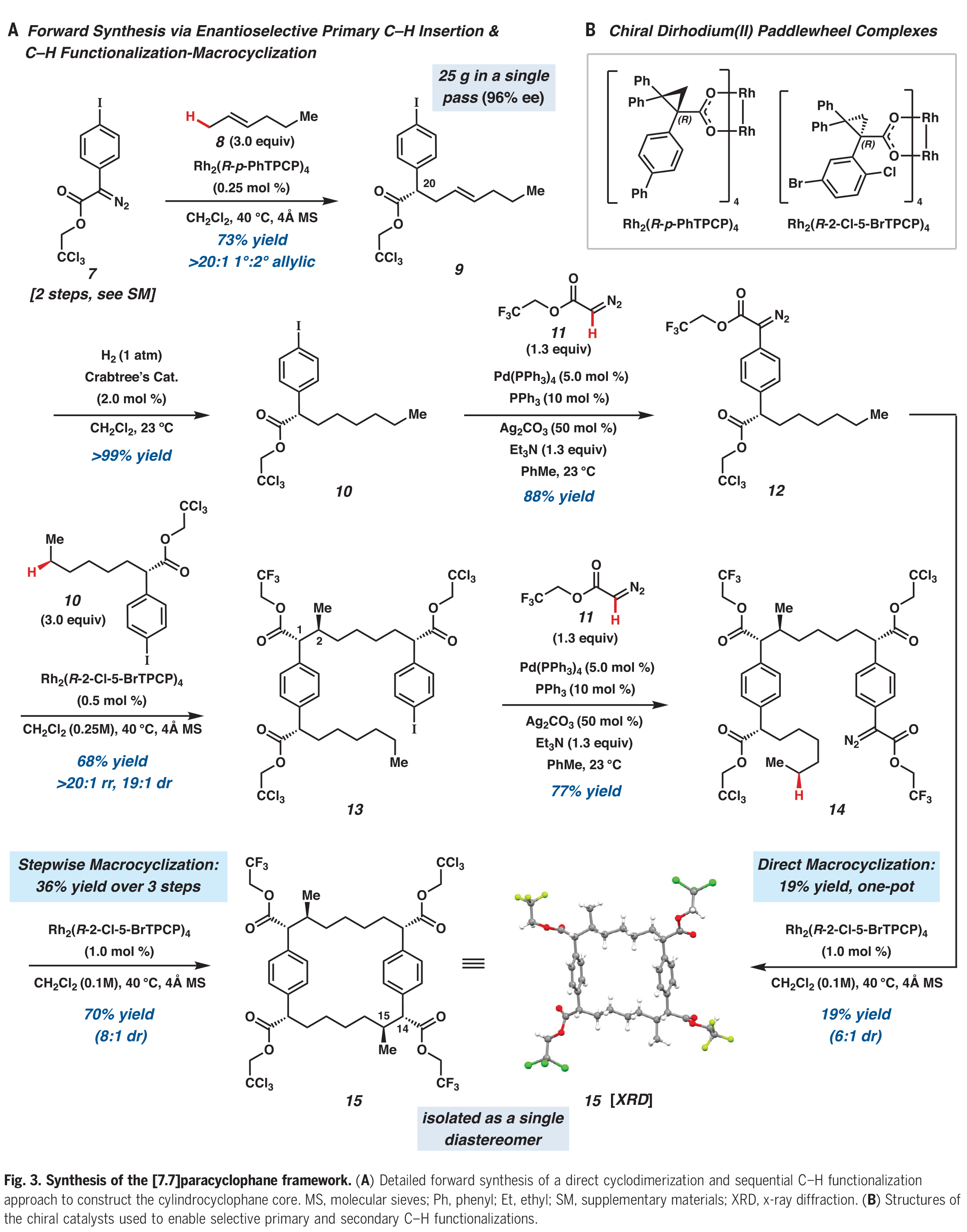
在构建了大环骨架后,大环15中的三氯乙酯被化学选择性地转化为双weinreb酰胺16(Fig. 4)。随后将剩余的三氟乙酯水解得到双羧酸17,再进行光催化的脱羧乙酰氧基化得到醋酸双苄酯3。在此阶段,作者进行了合成中最后的C-H官能团化反应,即大环3与的四个C(sp2)-H乙酰氧基化,这种转化只有在余金权小组所发展的反应条件下才能实现。在单次转化中,酰胺导向3的四个C(sp2)-H乙酰氧基化可以以60%的产率得到大环2,此转化标志着所有所需的C-H官能团化步骤的完成。
完成此合成的最后一个挑战是安装两个丙基侧链。作者发现通过酚类乙酸酯的化学选择性脱酰化,随后通过甲基化产生四甲基醚18。用DIBAL处理大环18可以使剩余的乙酸酯发生还原断裂,且两个Weinreb酰胺会被还原为相应的醛。这些醛可以直接进行Wittig烯烃化,以单一的烯烃异构体得到双烯烃19。最后,双烯烃19通过加氢得到20,并在Hoye课题组报道的条件下通过去甲基化得到(−)-cylindrocyclophane A。
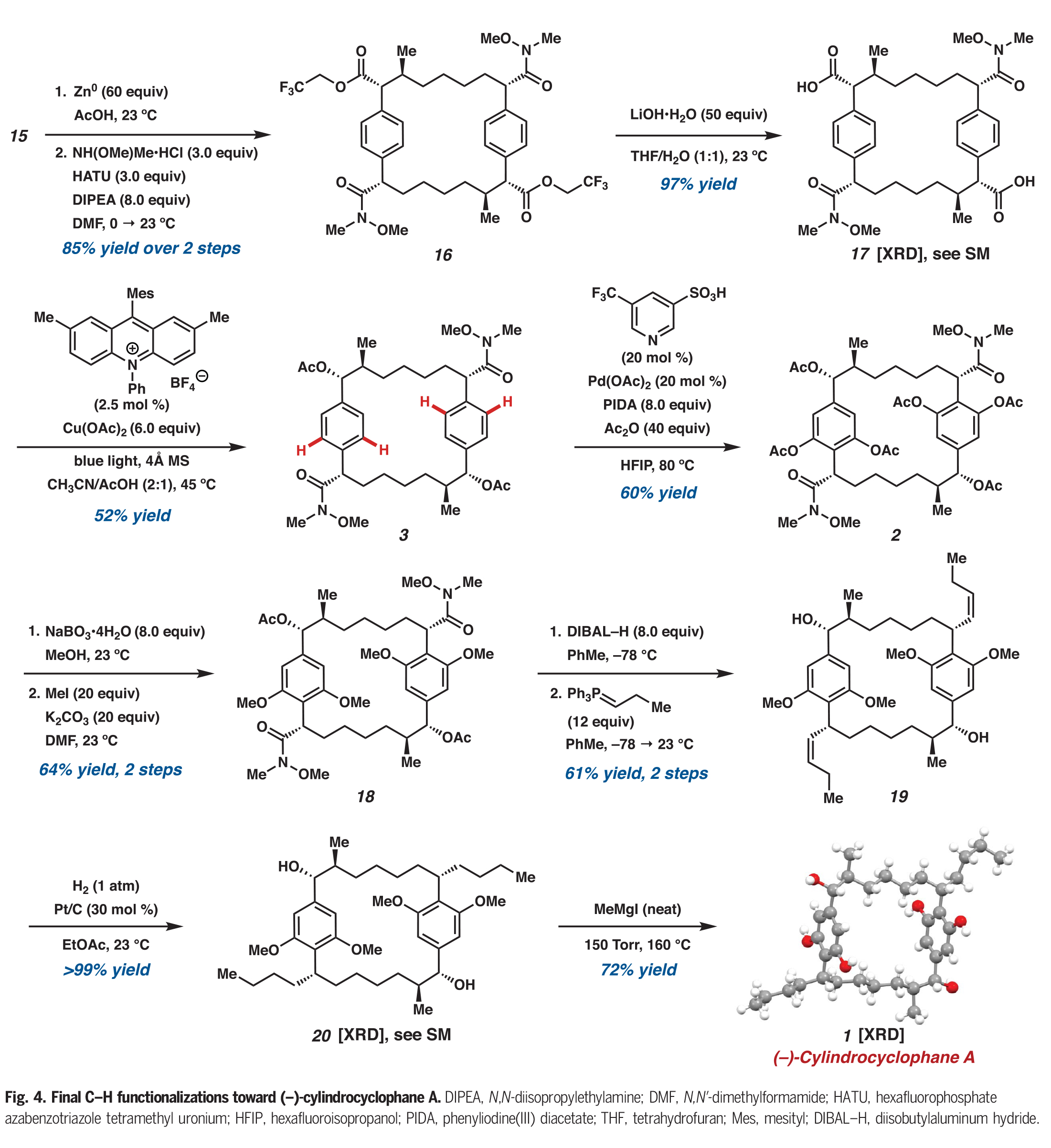
(图片来源:Science)
总结
Huw M. L. Davies课题组与Brian M. Stoltz课题组利用商业可得的起始原料,通过10个C-H官能团化反应形成了6根碳-碳键和4根碳-氧键,以良好的对映选择性和效率(17步)实现了(−)-cylindrocyclophane A的全合成。该路线展示了四种催化剂控制的对映选择性和非对映选择性C-H官能团化,两种钯催化重氮羰基化合物的C-H官能团化和四种酰胺导向的C-H乙酰氧基化构建了天然产物的所有六个立体中心。此项研究证明了C-H官能团化可以作为一种重要的合成手段,选择性地将低成本原料转化为高度官能团化和立体化学复杂的结构骨架。



 扫码下载APP
扫码下载APP
 科普中国APP
科普中国APP
 科普中国
科普中国
 科普中国
科普中国